
Canada Gazette, Part I, Volume 156, Number 51: Regulations Amending Certain Regulations Made Under the Food and Drugs Act (Agile Licensing)
December 17, 2022
Statutory authority
Food and Drugs Act
Sponsoring department
Department of Health
REGULATORY IMPACT ANALYSIS STATEMENT
(This statement is not part of the Regulations.)
Executive summary
Issues: The pace of innovation today means that drugs and medical devices are evolving more rapidly than the traditional regulatory frameworks that were designed to regulate them. In addition, there has been a shift among international regulators towards greater post-market oversight with respect to drugs and medical devices. Over time, Health Canada has been introducing legislative and regulatory amendments as well as implementing certain practices through policy to address these issues. Further regulatory amendments are required to provide a legal framework to support policy practices and to ensure transparency, predictability, consistency and compliance.
Many of the regulations specific to biologic drugs (biologics) are overly prescriptive, product-specific and do not reflect current science and technology. The prospect of removing product-specific requirements for biologics highlighted the need to clarify the existing quality control rules and expectations under good manufacturing practices (GMP) requirements that apply to all products.
Diverse subpopulations, such as women, racial minorities, children and the elderly, are often underrepresented in clinical trial data, impacting Health Canada’s ability to identify risk and the different safety and effectiveness profiles within diverse subpopulations.
Lastly, the current requirements do not allow manufacturers to qualify limits for purity and potency that are different from those in publications listed under Schedule B of the Food and Drugs Act (the Act) that could be considered acceptable by Health Canada when a manufacturer’s standard is claimed. Furthermore, manufacturers of certain drugs are required to indicate the standard on the label of their drug, which is a Canadian-specific requirement and at times has created challenges for manufacturers due to limited space available on the label.
Description: Health Canada is proposing new targeted provisions and regulatory amendments to the Food and Drug Regulations (FDR) and the Medical Devices Regulations (MDR) that would deliver on the Department’s modernization commitments and leverage long-standing policies and practices. The proposal would take into account recent experience with regulatory agilities successfully piloted through the COVID-19 interim orders and their transition to regulations. This proposal is comprised of distinct components that would
- Enable the use of terms and conditions (T&Cs) on the drug identification number of any drug;
- Broaden the scope of use of T&Cs for Class II, III, and IV medical devices;
- Require risk management plans (RMPs) for certain human drugs to manage risks and uncertainties;
- Allow for rolling reviews of certain drug submissions, including those for drugs intended to address a public health emergency;
- Clarify expectations that a drug be fabricated, packaged/labelled, tested and stored, including during transportation, in a manner that assures its quality;
- Modernize requirements for biologics by repealing outdated requirements and replacing them with those that reflect current practices;
- Clarify, in regulation, the authority to consider certain information obtained outside of a new drug submission to support Health Canada’s examination of that submission for a new drug;
- Require manufacturers to submit human clinical trial data broken down by population subgroups (disaggregated data) for new and supplemental human drug submissions, as submitted to the United States Food and Drug Administration (USFDA) or the European Medicines Agency (EMA); and
- Update requirements respecting standards for labelling and requirements for those that claim a manufacturer’s standard for their drug.
Rationale: For several years, Health Canada has been actively engaging in legislative and regulatory modernization to support a drug and medical device framework that would more effectively oversee regulated products over the entirety of their life cycle. The proposal seeks to deliver on the regulatory modernization commitments set out in the Health and Biosciences: Targeted Regulatory Review – Regulatory Roadmap, which supports the reduction of regulatory irritants and roadblocks to innovation by making Canada’s science-based regulatory system more agile and internationally aligned.
Through the inclusion of T&Cs, the Regulations Amending Certain Regulations Made Under the Food and Drugs Act (Agile Licensing) [the proposed regulations] would enable the Minister of Health (the Minister) to better manage risks and uncertainties with respect to drugs and medical devices, and to adapt to innovation and changes in therapeutic products. Formalizing into regulation Health Canada’s long-standing practice with respect to RMPs would better support the on-market evaluation of information that could have an impact on the benefit-risk profile of human drugs. The proposed amendments could facilitate earlier market access for those drugs that are eligible for a rolling review.
Quality control requirements would be clarified as they relate to all drugs to complement the proposed amendments for biologics. Product-specific requirements for biologics would be replaced with broader, more flexible regulations that would better address advancements in science and technology, and support current practice.
The proposed amendments would also clarify in regulation the Minister’s authority to consider information and material obtained from sources other than the submission, to support the review of a new drug, consistent with current practice.
Furthermore, access to disaggregated data as submitted to the USFDA or the EMA would incrementally enhance Health Canada’s ability to assess the safety and effectiveness of new drugs for human use in certain population subgroups. This step toward promoting increased diversity in clinical trials underscores Health Canada’s continued commitment to address the growing evidence of potential health disparities for equity-seeking and rights-holding populations, such as women, racial and ethnic minorities, or Indigenous Peoples, due to their underrepresentation in clinical data.
Finally, updating the regulations respecting labelling of the standard for specific drugs and requirements for certain drugs that claim a manufacturer’s standard would address long-standing concerns from manufacturers that the current requirements are not always necessary to ensure the safety, effectiveness and quality of a drug.
Cost-benefit statement: The incremental costs for industry are estimated at $158 million (present value [PV]). It is expected to cost Health Canada $26 million (PV) to review and manage T&Cs, rolling reviews and RMPs. As a result, the total anticipated cost of the proposed amendment is $184 million (PV) over a 10-year period, discounted at 7%.
These costs would be offset by earlier market authorization, an improvement in Canadians’ quality of life, and the elimination of regulatory requirements around standards. These benefits are estimated at $440 million (PV) over a 10-year period. The total anticipated net impact of this proposal is $256 million (PV) in net benefits.
One-for-one rule and small business lens: This proposed amendment is an out for the purpose of the one-for-one rule as the anticipated administrative burden to the industry is estimated to be reduced by $25,177 (2012 dollars) annually, or $1,678 (2012 dollars) per business. The small business lens applies as there are approximately 225 small businesses in Canada that may be affected by this proposal.
Issues
The pace of innovation today means that drugs and medical devices are evolving more rapidly than the traditional regulatory frameworks that were designed to accommodate them. As a result, the Minister has limited ability to compel manufacturers to take steps to manage the risks and uncertainties associated with a drug or medical device, or to accommodate different manners in which information relating to drug submissions could be provided or obtained. While Health Canada has implemented certain practices through policy to address these issues, there is a need to codify such practices and promote compliance through measures that provide transparency, predictability and consistency.
With regard to the fabrication, packaging/labelling, testing, storing and transportation of a drug, despite the FDR assigning quality control departments responsibilities with respect to procedures and methods, there is no clear requirement that such activities must be conducted in a way that maintains and assures the quality of the drug. One such example of the existing gaps is the expectation that drugs should be stored according to their approved storage conditions.
In addition, many of the provisions related to biologics do not reflect the current science, technology, and ways in which the industry has evolved over the last 70 years. Many of the provisions are overly prescriptive, product-specific, and are no longer scientifically relevant. This presents a risk to the health and safety of Canadians, which Health Canada currently addresses through flexible and outcome-based practices and policies.
When examining a drug submission, in order for the Minister to make the most informed decision about the authorization of the drug, the Minister may consider information that could come from other sources than the submission. Currently, the sources of information that are explicitly permitted by regulation are too limited and do not include key sources of material and information that may be needed for Health Canada to assess and authorize a new drug.
Additionally, Health Canada does not currently have a requirement to receive disaggregated data to support the evaluation of submissions for new drugs or supplements to new drug submissions. However, research and evidence suggest there can be different effectiveness and safety profiles for some drugs within diverse subpopulations, including those that are frequently underrepresented in clinical trials (e.g. women, racial minorities, children and the elderly). Receiving disaggregated data would allow Health Canada the opportunity to better evaluate a drug’s safety and effectiveness within diverse subpopulations and identify whether it may pose an increased risk for a certain subpopulation as compared to others.
Finally, where a manufacturer’s standard is claimed, the FDR do not allow manufacturers of drugs to set limits for purity and/or potency that are different from those in publications listed under Schedule B of the Food and Drugs Act (the Act) with additional data that could be considered acceptable to Health Canada. Manufacturers have indicated that this requirement has occasionally resulted in a drug being pulled off the market in Canada. Furthermore, labelling requirements pertaining to the standard of a drug are Canadian specific (i.e. not required by other regulators, such as the USFDA and EMA) and at times have created challenges for some manufacturers due to limited space available on the label.
Background
The Minister of Health is responsible for regulatory activities related to the safety, effectiveness and quality of drugs and medical devices. The Minister’s authority is derived from the Act and regulations made under it, including the FDR and the MDR.
For several years, Health Canada has been actively engaging in legislative and regulatory modernization to support drug and medical device frameworks that effectively oversee regulated products over the entirety of their life cycle. In 2014, Parliament adopted Vanessa’s Law, which amended the Act to include regulation-making authorities for the purposes of gathering safety information respecting drugs and medical devices, also known as therapeutic products, and authorities that enable the Minister to take action in the event that a serious health risk is identified. This was a key step in Health Canada’s initiative to modernize the Act and its associated regulations respecting therapeutic products. Regulations made under the new Vanessa’s Law authorities have introduced measures that have
- Strengthened the safety oversight of therapeutic products throughout their life cycle (e.g. T&Cs for opioids and designated COVID-19 drugs);
- Improved post-market surveillance (e.g. mandatory reporting of serious drug reactions and medical device incidents by hospitals, reporting of foreign regulatory actions involving serious risk to human health);
- Expanded post-market authorities for medical devices;
- Promoted greater confidence in the oversight of therapeutic products by increasing transparency (e.g. public release of clinical trial information); and
- Aligned requirements with regulatory authorities from other jurisdictions.
Further work on identifying and responding to regulatory irritants and roadblocks to innovation was conducted as part of the 2019 Health and Biosciences Sectoral Regulatory Review, and resulted in the launch of Health Canada’s Regulatory Innovation Agenda. Through these initiatives, Health Canada has committed to modernizing applicable regulations to ultimately deliver modern, flexible regulatory frameworks for drugs and medical devices.
Some of these flexibilities (T&Cs and rolling reviews) were piloted as part of the response to the COVID-19 pandemic through the following regulatory packages:
- The Interim Order Respecting the Importation, Sale and Advertising of Drugs for Use in Relation to COVID-19 (September 2020), followed by amendments to the FDR (March 2021); and
- Three Interim Orders Respecting the Importation and Sale of Medical Devices for Use in Relation to COVID-19 (March 2020, March 2021, February 2022).
This regulatory initiative is part of Health Canada’s Regulatory Innovation Agenda and will contribute directly to the Government’s Biomanufacturing and Life Sciences Strategy that recognizes the importance of the regulatory system as an enabler of the growth of the domestic biomanufacturing sector.
Terms and conditions
The rapid pace of innovation in industry has led to uncertainties and risks in relation to therapeutic products that may not be adequately managed through existing regulatory provisions. A T&C obligation is one that the Minister may impose on the holder of a therapeutic product authorization to conduct an activity with respect to the drug or medical device to which the authorization applies. The main purpose of T&Cs is to optimize the benefits and manage risks and uncertainties associated with the drug or medical device, including by collecting additional information after it has been authorized. Health Canada has long recognized the value of such an approach. In 1998, the Notice of Compliance with Conditions (NOC/c) policy was introduced to enable manufacturers of certain human drugs — namely, those intended to treat a serious, life-threatening or severely debilitating disease or condition — to apply for an authorization to sell a drug based on promising clinical evidence that indicates its use is reasonably likely to produce the intended result. This policy has allowed Health Canada to more effectively manage uncertainties related to risks and benefits and, as a result, facilitate earlier market access for potentially life-saving drugs when the manufacturer agrees to undertake further studies to confirm the effectiveness and monitor the safety of those drugs and provide that information to the Minister following its entry onto the Canadian market.
Currently, the Minister has a broad power to impose or amend T&Cs on opioid drugs and designated COVID-19 drugs in the FDR. Since the MDR came into force in 1998, the Minister was granted the authority to impose or amend T&Cs related to testing on Class II, III and IV medical device licences. Further, broad T&C powers were included in recent interim orders related to the authorization of COVID-19 medical devices.
Risk management plans
An RMP summarizes the identified and potential risks and uncertainties related to a drug and the associated pharmacovigilance activities, as well as other measures that the manufacturer intends to put in place to manage those risks and uncertainties.
Internationally, most regulators of human drugs, including those in the European Union, the United Kingdom, and the United States, have introduced legal requirements for RMPs or their equivalent. Since 2009, to better align with international practice, Health Canada has requested RMPs from manufacturers, on a voluntary basis, upon filing of a new drug submission for some new active substances.footnote 1 In 2015, Health Canada published the Guidance Document - Submission of Risk Management Plans and Follow-up Commitments that formalized the practice of requesting RMPs from manufacturers of
- Pharmaceutical drugs (which include prescription and non-prescription pharmaceutical drugs) that include new active substances;
- Biologics (which include biotechnology products, vaccines and fractionated blood products) as set out in Schedule D to the Act; and
- Radiopharmaceutical drugs as set out in Schedule C to the Act.
The guidance document indicates that manufacturers should include Canadian-specific sections that would discuss factors that are applicable to the Canadian population, as applicable to the Canadian context and marketplace.
Rolling reviews
A “rolling review” allows a manufacturer to file its drug submission with some but not all of the information necessary for the regulator to assess the safety and effectiveness of a drug. Provided certain conditions are met, such a submission may be filed with the understanding that the missing information would need to be provided within a reasonable amount of time. However, regardless of how much information is included when the submission is filed, the decision to authorize the drug can only be made when all of the required information, including the missing information, has been provided and found to be acceptable.
Health Canada has used rolling reviews to increase timely access to needed drugs. Currently, rolling reviews are available by practice for annual updates of seasonal influenza vaccines as well as for simultaneous review of veterinary drugs with the United States under the Canada–United States Regulatory Cooperation Council. Rolling reviews are also available for new drug submissions for designated COVID-19 drugs as set out in the FDR as amended by the Regulations Amending the Food and Drugs Regulations (Interim Order Respecting the Importation, Sale and Advertising of Drugs for Use in Relation to COVID-19).
Assuring drug quality during manufacturing
Good manufacturing practices are an internationally recognized quality assurance system used to ensure that drugs are consistently fabricated, packaged, labelled, tested, imported, distributed and wholesaled. However, while it is expected that drugs are fabricated, packaged, labelled, tested and stored, including during transportation, in a manner that would assure the quality of the active ingredient and the drug in dosage form, this is not clearly stated in regulation.
Modernizing requirements for biologics
Biologics are manufactured and extracted from living tissue or organisms, some with modified genes. Their safety and effectiveness are highly dependent on their source materials (e.g. cells, living organisms) and auxiliary materials (e.g. additives used to supplement cell culture medium). In addition, biologics tend to be sterile, injectable solutions that treat serious or life-threatening conditions, and the risks related to these drugs can be equally serious or life-threatening.
Much of Part C, Division 4, of the FDR consists of product-specific regulations that were introduced in the 1950s and 1960s to respond to issues of the day. As the number of biologics has greatly increased over time and since regulations have historically been largely product-focused, they have not kept pace with scientific advances.
Significant amendments were made in 1997 in order to amalgamate requirements for establishment licences for the manufacturing of pharmaceutical drugs, biologics, and radiopharmaceutical drugs into Division 1A of the FDR. While these changes introduced GMP inspections related to the premises, personnel, process, and products, Health Canada maintained by policy the ability to use information obtained on site to confirm the manufacturer’s ability to consistently produce a safe biologic and verify information provided in the related submission.
Information considered to support the examination of drug submissions
In line with the above-mentioned practice, Health Canada uses information from a variety of sources, including On-Site Evaluations (OSEs) and GMP inspections, in the examination of a drug submission, for the purpose of assessing the safety and effectiveness of a drug. Health Canada currently considers information and material available, including that obtained outside the submission, to make an informed decision about whether to authorize a drug for the Canadian market.
Disaggregated clinical trial data for new human drug submissions and supplemental new human drug submissions
The collection of disaggregated data about clinical trial participants is a key measure Health Canada can take to first assess and, if needed, address any discrepant risks and benefits of drugs in diverse subpopulations. Health Canada places importance on the development and conduct of inclusive clinical trials particularly for underrepresented populations. Applications to Health Canada generally have some degree of disaggregated population data; however, it is not always fully reflective of the patient population or disaggregated by population subgroups (e.g. age, sex, gender or race).
Standards
Drug standards include criteria and test methods that help to assure the quality of a drug. These standards can be prescribed in regulation, set out in a pharmacopoeiafootnote 2 listed under Schedule B of the Act or developed by the manufacturer of the drug. If a standard has not been prescribed in regulation, but it appears in a pharmacopoeia listed under Schedule B of the Act, a manufacturer may use either the pharmacopoeial standard or their own manufacturer’s standard. If a manufacturer’s standard is used, the FDR require that the drug meet the highest degree of purity and the least variation in potency for that drug of any pharmacopoeias listed under Schedule B. This applies to all drugs, including active ingredients, regulated under Part C, Division 1, 3, 4 or 8, of the FDR throughout their life cycle. This prescriptive requirement does not allow those claiming a manufacturer’s standard to scientifically justify wider controls for purity and potency that may also be acceptable to the Minister.
For drugs regulated under Division 1 or 8 of the FDR, the Regulations currently require that the inner and outer labels indicate if a standard prescribed in the FDR, Schedule B standard (e.g. U.S. Pharmacopeia) or manufacturer’s standard has been used. The inclusion of this information is a Canadian-specific requirement and at times has created challenges for manufacturers due to the limited space available on the label.
Objective
The objective of the proposed regulatory amendments is to increase regulatory agility to keep pace with innovation and facilitate access to advanced treatments and promising therapies, while continuing to ensure authorized drugs and licensed medical devices are safe, effective, and subject to appropriate oversight.
Description
These proposed regulatory amendments would introduce a series of targeted changes to the FDR and the MDR that are reflective of existing policies as well as Health Canada’s recent experiences with the COVID-19 interim orders.
Terms and conditions — Drugs and medical devices (amendments to the FDR and MDR)
The proposed amendments would allow the Minister to impose T&Cs on a drug identification number of any drug or on a Class II, III or IV medical device licence at the time it is issued, or at any time later. T&Cs could also be amended or removed at any time if necessary. Although the MDR currently allow the Minister to impose T&Cs related to testing on Class II, III or IV medical device licences, the proposed amendments would broaden the scope of T&Cs that can be imposed and allow them to be enforceable under section 21.7 of the Act.
Before imposing or amending T&Cs, the Minister would consider whether the T&Cs contribute to meeting the objectives of managing uncertainties related to the benefits and risks, optimizing the benefits and managing the risks, and identifying changes related to the benefits and risks of the drug or the medical device.
Furthermore, the Minister would consider whether the T&Cs are technically feasible, whether there are less burdensome means of achieving those objectives, and whether they could be achieved through the application of existing requirements under the Act or the FDR and the MDR, as the case may be.
All drug submissions must have the necessary information and data to establish the safety, efficacy and quality of a drug to demonstrate a favourable benefit/risk profile and satisfy all requirements under the FDR for market authorization. The overall objective of imposing T&Cs would be to ensure a drug maintains a favourable benefit/risk profile throughout its life cycle and that the safety, efficacy, or quality of a drug has not changed from when the market authorization was issued.
T&Cs could, for example, be applied to
- Manage the uncertainties relating to the benefits and/or risks identified at the time they are issued (e.g. request studies to confirm benefits of a drug or device, including in subpopulations, if necessary);
- Manage emerging risks or uncertainties that have surfaced post-market and may not have been identified prior to authorization;
- Collect data to be able to identify and assess potential changes to safety and/or effectiveness and to manage uncertainties (e.g. for new technologies or new indications for use); or
- Require the medical device licence holder to make changes to the medical device to continue to maintain safety and effectiveness.
Initially, in the case of drugs, the proposed amendments would be limited to any public health emergency drug, which would be a drug that relates to COVID-19 or to a condition described in the List of Conditions that Threaten Public Health in Canada, which would be incorporated by reference. However, one year after registration of the proposed regulations, those limited provisions would be repealed and the proposed amendments respecting T&Cs for all drugs would come into force. The proposed amendments respecting T&Cs for Class II, III and IV medical devices would also come into force at that time.
Risk management plans (amendments to the FDR)
The proposed amendments would reflect Health Canada’s long-standing practice set out in the guidance document entitled Submission of Risk Management Plans and Follow-up Commitments. The regulations would require an RMP to be filed with an application for a drug identification number, a new drug submission or an abbreviated new drug submission for a human drug if the Minister has reasonable grounds to believe that there is a significant degree of uncertainty respecting the risks associated with the drug, or if the drug presents a serious risk of injury to human health that warrants measures beyond changes to the label, to reduce the probability or severity of such an injury. RMPs would be required for all extraordinary use new drug submissions.
The amendments would also provide the Minister with the regulatory authority to require an RMP for an authorized human drug for which an RMP has not yet been provided where the circumstances described above arise.
In addition, the authorization holder would be required to provide the Minister with an updated RMP if the updated RMP is significantly different from the previously submitted RMP, or if the Minister has reasonable grounds to believe that an updated RMP is required to respond to significant differences in
- The risks and uncertainties associated with the drug; or
- The measures warranted to manage a serious risk of injury to human health.
To support regulatory transparency, the amendments would also create a new obligation on manufacturers to submit a summary of new and updated RMPs in both official languages. In practice, these summaries would be published online and their contents would be aligned with the content of other jurisdictions, with the addition of Canadian-specific content required when relevant.
The proposed amendments related to RMPs would not apply to drugs for veterinary use.
Rolling reviews (amendments to the FDR)
The proposed amendments would introduce a number of rolling review options into regulation for new drug submissions and supplementary new drug submissions where
- The new drug meets certain conditions as described below;
- The drug is a veterinary drug and the Minister has indicated an intent to conduct the review with a foreign regulatory authority;
- There has been a change to the strain to which an influenza vaccine, referred to in a list incorporated by reference, relates; or
- The drug relates to a condition that threatens public health set out on a list incorporated by reference.
These options all provide manufacturers seeking a notice of compliance (NOC) with modified requirements for the submission of information at the time of filing. Any missing information needed to assess the safety and effectiveness of the drug must be provided within a reasonable time to allow Health Canada to make a determination about issuing an NOC. All other requirements under the FDR, such as record retention and data protection, as well as the intellectual property regimes under the Patent Act, would also apply to those submissions and drugs that are subject to a rolling review.
Drugs that meet certain conditions
The proposed regulations would create a new optional process for new drug submissions and for supplements to new drug submissions to facilitate timely access to new drugs that meet one or more of the following conditions:
- Drugs for human and veterinary use that are needed to address emerging infectious diseases that pose or may pose a serious risk of injury to health; and
- Drugs for human and veterinary use for the treatment, prevention, mitigation or diagnosis of serious diseases or conditions where
- The recommended purpose and conditions of use of the new drug do not fall within the recommended purpose and conditions of use of any other drug for which a drug identification number has been assigned and has not been cancelled, or
- The new drug is significantly more effective, or possesses a significantly lower risk, compared to every other drug for the same recommended purpose and conditions of use for which a drug identification number has been assigned and has not been cancelled.
A pre-submission assessment to determine eligibility for a rolling review where the new drug meets one or more of the above conditions would require that the manufacturer
- Provide Health Canada with information that demonstrates that the proposed drug meets one or more of the conditions described above and that the manufacturer possesses a significant amount of evidence to establish the safety and the clinical effectiveness of the drug for the purpose and conditions of use recommended; and
- Provide a plan that sets out how and when, within 155 days from when the submission is filed, they will submit any missing information. The plan is expected to contemplate providing the missing information in a few transactions.
If it is determined that the proposed drug submission, for which a pre-submission assessment was conducted, is eligible for a rolling review, the manufacturer would be notified by the Minister. The notice would indicate a deadline of 60 days after the day on which the notice is issued by the Minister to provide the submission, as well as how and when the missing information is to be provided.
In order to allow Health Canada to begin the review of these drug submissions, the proposed amendments would also set out minimum information requirements that must be met when the submission is filed, including that the submission must, at the time of filing, include a significant amount of information to enable the Minister to begin to assess the safety and effectiveness of the drug. At the time of filing, a drug submission that is the subject of a rolling review must also include all of the information that is normally required respecting the formulation, medicinal ingredient, use, and dosage form of the drug.
With respect to a new drug submission, the information that would be allowed to be provided following the initial transaction would include some missing information respecting the tests and evidence to establish the safety and effectiveness of the new drug, information respecting the investigators to whom the new drug has been sold, evidence that test batches of the new drug were manufactured in a way that is representative of market production, the withdrawal period of the new drug in food-producing animals, a risk management plan, and any summaries and sectional reports for any studies that have yet to be completed. With respect to a supplement to a new drug submission, which is limited to matters that are significantly different compared to the original submission, the rolling option would require a substantial amount of information to be provided in the initial transaction. However, this option would allow for the subsequent submission of any missing information the Minister needs to assess the safety and effectiveness of the new drug, as well as any summaries and sectional reports for any studies that have yet to be completed.
All of the information that is normally required respecting a drug submission would have to be provided before the Minister can complete the examination of the submission. The filing date for a submission that is eligible for a rolling review would be the date that the submission is determined to be administratively complete by Health Canada (i.e. once all the elements and forms required for processing are completed and have been submitted). This date may differ from the date of receipt of the initial transaction, should the submission be considered to be administratively incomplete at that time. Once established, the filing date does not change, even though, in the case of a rolling review, the manufacturer provides Health Canada with missing information afterwards.
Drugs already subject to a rolling review per current policy
In addition to the above-noted rolling review options, in keeping with current policy, veterinary drug submissions where the Minister has indicated their intent to conduct the review with a foreign regulatory authority, and supplementary new drug submissions where there has been a change to the strain to which an influenza vaccine relates (e.g. submissions respecting annual updates to seasonal influenza vaccines), would also be eligible for a rolling review. Influenza vaccines eligible for this rolling review option will be set out in a list incorporated by reference. This list would be developed, reviewed and maintained in accordance with the guiding principles set out in Health Canada’s Incorporation by Reference Policy. In order to align with current practice, requirements for both of these rolling review options would differ. For example, these submissions would not be subject to a pre-submission assessment.
With the exception of rolling reviews for submissions where there has been a change to the strain to which an influenza vaccine relates, the proposed amendments for the above-described rolling review options would also allow the Minister, under certain conditions, to cancel the submission. For example, in the case of rolling reviews for new drugs that meet the conditions described in the section above, where the manufacturer had failed, or would be unable, to provide the missing information within a reasonable period of time after the relevant date specified in the notice, the review would be terminated and the submission would be considered to be cancelled by the manufacturer. If a manufacturer chooses to file a new submission for the same drug with Health Canada after such a cancellation, a new filing date would be assigned once the new submission is determined to be administratively complete.
Public health emergency drugs
Finally, existing provisions in the FDR respecting licensable activities under Division 1A related to COVID-19 and the market authorization of COVID-19 drugs, including the option for a rolling review, would be expanded to apply to any public health emergency drug in relation to a condition referred to on the List of Conditions that Threaten Public Health, which would be incorporated by reference. A public health emergency drug would be defined to mean a new drug for which the purpose and conditions of use recommended by the manufacturer relate to COVID-19 or a condition that is described on this list. In order to add conditions to this list, the Minister would need to have reasonable grounds to believe that the condition presents, or is the result of, a significant risk to public health in Canada, and immediate action is required to deal with the risk. The proposal to introduce a list that would be incorporated by reference would allow the Minister to respond to a public health emergency in a manner that is timely and flexible. The list would be developed, reviewed and maintained in accordance with the guiding principles set out in Health Canada’s Incorporation by Reference Policy.
Supporting the extension of the existing COVID-19 rolling review option to public health emergencies, the criteria for pre-positioning a drug would also be expanded to include a public health emergency drug to allow a response to a condition referred to on the list. The regulations would also be amended to allow the importation only if the holder of the drug establishment licence is an authorized importer and the public health emergency drug is within the same category of drugs as is authorized by the licence.
Assuring drug quality during manufacturing (amendments to the FDR)
The proposed amendments would clarify expectations that a drug be fabricated, packaged/labelled, tested, and stored, including storage during transportation, in a manner that assures the drug’s quality.
This requirement is meant to apply to all licensed fabricators, packagers/labellers, testers, and wholesalers, as well as Canadian importers and distributors. The new provision C.02.012.1 is intended to work together with other requirements of Division 2 of the FDR and does not change the existing expectations for all drugs.
Modernizing requirements for biologic drugs (amendments to the FDR)
The modernization of Part C, Division 4, of the FDR proposes to remove outdated product-specific regulations for biologics, and replace them with more general requirements that reflect the underlying safety purposes, as they are applied through current guidance and practice. The provisions proposed to be removed and replaced are the product-specific provisions spanning between C.04.050 and C.04.683. As a result, the definitions “date of manufacture” and “date of issue” would no longer be required and therefore would not appear in the new Division 4.
The proposed modernization would be implemented in a manner that minimizes impact on manufacturers of currently marketed biologics by recognizing and supporting current practices. More specifically, the proposed amendments to the FDR are not expected to change existing practices, including those with regard to lot release and OSEs.
Controls over manufacturing of biologics
The proposed amendments reflect current practice and would replace the prescriptive requirements for biological starting and auxiliary materials used in the manufacturing of biologics with more flexible, outcome-based regulations that maintain an appropriate level of safety oversight. Biological source materials have been defined to include biological raw materials, biological starting materials and auxiliary materials. The requirements regarding the fabrication and collection of biologics have been clarified to also apply to biological source materials.
Existing regulations controlling contamination of biologics are proposed to be generalized to ensure that production personnel do not contribute to the risk of contamination of the biological source materials by infectious agents. The proposed amendments would also remove certain personnel, storage and transportation requirement provisions as they are outdated, and the Department would instead rely on corresponding regulations under Part C, Division 2 (Good Manufacturing Practices), of the FDR.
Standards for biologics
Individual standards for biologics, such as insulin, that are prescribed in Division 4 are out of date with changes in science and are not currently relied upon by manufacturers or Health Canada. The intent is to replace these prescribed standards and continue to assess specifications provided by the manufacturer during the review of the submission. In addition, the proposed amendments would require that reference preparations used to evaluate the purity or potency of a drug, as applicable, be adequate to control the quality of the product.
Lot release of biologics
Provisions supporting the lot release program would be amended to better support a risk-based, tiered approach, as per Guidance for Sponsors: Lot Release Program for Schedule D (Biologic) Drugs and current practice.
To manage risks associated with individual lots, the proposed amendments would enable the Minister to ask for information, samples or other materials, as required, including for conducting independent testing. Where a request has been made, no person would be permitted to sell any drug from that lot until the Minister notifies them that the lot may be sold.
The proposed amendments would also formalize into regulation the current discretionary practice of providing yearly biologic product reports. These reports characterize the quality of the drug and its active ingredients, and the consistency of their manufacturing and packaging processes over the period of time since the last report. The proposed requirements include periodic quality reporting on an annual basis or longer as specified by the Minister.
Labelling of biologics
The proposed amendments would update some labelling requirements for biologics to align them with the requirements under Part C, Division 1, of the FDR. These include labelling flexibilities for small containers that have an outer label.
In addition to adequate directions for use, the proposed regulations would require on the label a statement indicating the approved storage conditions, and additional information when necessary to prevent injury to health of the consumer/patient (multi-dose, pediatric use, age group, warning, etc.).
The labels of biologics would be required to indicate if the drug is derived directly from a human source or an animal source and species of origin.
The proposed amendments would allow manufacturers of biologics that are stockpiled by government departments or agencies for use in emergency situations, where the drug is anticipated to be stored for prolonged periods and the long-term stability program is ongoing, to indicate the expiry date by other means than through a statement on the label on the package. The alternate means of indicating the expiry date would have to be readily available to the person administering the drug, and be acceptable to the Minister.
All of these proposed labelling amendments are in line with current practices and therefore are not expected to impact approved labels.
Information considered to support the examination of drug submissions (amendments to the FDR)
In line with current practice, the proposed amendment to provision C.08.003.1 would clarify the Minister’s authority to consider information or material that could be examined on a risk-based, case-by-case basis during Health Canada’s assessment of a drug submission. The Minister could examine information and material
- Provided to the Minister by any person under the Act;
- Obtained from a building or site where a drug is manufactured, packaged, labelled or tested (e.g. on-site evaluations for biologics, GMP inspections); and
- Obtained from a foreign regulatory authority (e.g. foreign review reports, foreign inspection reports).
The updated provision is not meant to alter the manufacturer’s obligation to provide sufficient information to support the submission. The data protection provisions of the FDR and the Patented Medicines (Notice of Compliance) Regulations would continue to apply, even where information is considered under provision C.08.003.1.
Disaggregated clinical trial data for new human drug submissions and supplemental new human drug submissions (amendments to the FDR)
The proposed amendments would require manufacturers to submit human clinical data broken down into population subgroups to support the safety and effectiveness of the new (or supplemental) drug submission, if the disaggregated data has already been submitted to the USFDA or the EMA. This is an important step as Health Canada moves forward with an incremental approach towards additional data disaggregation requirements.
Standards (amendments to the FDR)
The proposed regulatory exemption respecting standards would exclude new drugs that are regulated under Part C, Division 8 of the FDR, other than those under Schedule C (radiopharmaceutical drugs), from having to meet the tightest standards for purity and potency of all the Schedule B pharmacopoeias in which the active ingredient or drug appears, where the manufacturer claims a manufacturer’s standard. Due to their unique intrinsic characteristics along with their sensitive biodistribution profile that ultimately impacts on their therapeutic and diagnostic index, Schedule C drugs would continue to be required to meet the tightest standards for purity and potency of all Schedule B pharmacopoeias when a manufacturer’s standard is claimed.
Drugs that are not new drugs would continue to be subject to the current requirements.
For drugs regulated solely under Division 1 and those regulated under Division 8 that are not also regulated under Divisions 3 and 4 (i.e. radiopharmaceutical and biologic drugs), the proposed amendments would remove the requirement for the standard used for the drug to be indicated on the package label.
Coming into force
The following proposed regulatory amendments that prioritize burden reduction for industry and ensure that Health Canada is well positioned to address any future public health emergencies would come into force upon registration:
- Amendments related to public health emergency drugs (including T&Cs, rolling reviews and pre-positioning);
- Assuring drug quality during manufacturing;
- Modernizing requirements for biologic drugs;
- Information considered to support the examination of drug submissions;
- Disaggregated clinical trial data for new human drug submissions and supplemental new human drug submissions; and
- Standards.
The following proposed regulatory amendments that require additional time to operationalize would come into force one year after registration:
- T&Cs (for all drugs and for Class II, III or IV medical devices);
- Risk management plans; and
- Rolling reviews (for drugs other than public health emergency drugs).
The following proposed regulatory amendments would come into force at a time set out in a future regulatory amendment once the Department determines that provisions specific to COVID-19 are no longer required:
- Amending the definition of public health emergency drug to no longer include a new drug for which the purpose or conditions of use recommended by the manufacturer relate to COVID-19; and
- Removing other COVID-19-specific provisions.
Regulatory development
Consultation
Previous consultations
The existing guidelines and policies, which form the basis for this proposal, were consulted on prior to their implementation and are now current practice. For example, Health Canada’s policy approach to RMPs was established based on the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use E2E guidelines (PDF). These guidelines were based on input received from the international pharmaceutical industry. Prior to implementing Health Canada’s final RMP guidance, which was well received by industry, significant consultation was done to ensure alignment with international guidelines that have been implemented in other jurisdictions.
Health Canada also heard from stakeholders on the need for regulatory agility during the 2018 Health and Biosciences Targeted Regulatory Review process.
Additionally, consultations were held before bringing forward the interim orders and regulations addressing COVID-19. The outcomes of these consultations are summarized in a What was heard report posted online, as well as in the medical devices interim orders explanatory notes and the regulatory impact analysis statement accompanying the regulations which transitioned the drug interim order into regulation. Stakeholders did not raise any concerns about the ability of the Minister to impose T&Cs on COVID-19 drugs and devices. Stakeholders were also supportive of the use of rolling reviews, with one stakeholder suggesting that rolling reviews be expanded to other life-saving drugs.
Flexibility in the labelling requirements respecting drug standards has been requested by manufacturers over the years. In addition, when claiming a manufacturer’s standard, manufacturers have indicated that the current requirements are overly restrictive respecting the purity and potency of a drug.
Stakeholders have expressed support for the modernization of the requirements for biologics through the years. The Department has completed significant policy and scientific technical analysis of both Division 4 and Schedule D for over 10 years. Throughout this analysis, industry stakeholders were consulted and have been supportive of Health Canada’s modernization efforts.
Notice of intent
Stakeholder feedback on some of the components was requested following publication of a 90-day notice of intent in the Canada Gazette, Part I. The consultation ran from July 31, 2021, to October 28, 2021. During this time, industry stakeholders were also asked to complete a cost-benefit survey. Meetings were also held with health technology assessment (HTA) organizations and health system partners such as the provinces and territories. These meetings were used to provide an overview of the notice of intent and answer any questions stakeholders had about the proposed changes to the FDR and MDR.
Response
Health Canada received 25 responses from pharmaceutical and medical device companies, industry associations, HTA organizations, provinces and territories, associations representing health care professionals and individuals, as well as 32 completed cost-benefit surveys from industry stakeholders. The feedback received has been used to refine the proposed amendments included in this proposal.
Stakeholders were generally supportive of the proposed amendments; however, they did request that additional details respecting implementation be provided. They expressed particular support for Health Canada’s efforts to increase regulatory agility, reduce known irritants, improve access to therapeutic products, and enhance post-market oversight throughout the life cycle of a drug or device.
Terms and conditions — Drugs and medical devices (amendments to the FDR and MDR)
With respect to T&Cs, some industry stakeholders recommended that T&Cs for drugs and medical devices should only be used in exceptional circumstances. HTA organizations wanted to better understand the implications of T&Cs on their review processes. Respondents also indicated that T&Cs should be harmonized internationally to reduce the burden and therefore cost to industry, and that an opportunity to be heard should be provided to the manufacturer before T&Cs are applied to a drug identification number. One association representing pharmacists advised that T&Cs for drugs should not result in lesser pre-market requirements and that care should be taken to not impact the practice of health care professionals. One medical device stakeholder questioned the need for T&Cs for Class II (i.e. lower risk) devices. An industry stakeholder indicated that it would be judicious to align T&Cs requirements for drugs and devices given that there are health products at the drug-medical device interface. To address these concerns, the proposed amendments to the FDR and MDR outline considerations that the Minister would have to take into account before imposing T&Cs. T&Cs would not be used as a mechanism to replace pre-market requirements.
RMPs (amendments to the FDR)
Some stakeholders expressed concerns about misrepresentation if Health Canada were to draft and translate summaries of their RMPs. In response to this concern, the proposed regulatory amendments would require that manufacturers include summaries of their RMPs in both official languages at the time of submission for review by Health Canada.
Stakeholders expressed a desire to maintain certain existing operational practices, such as the ability to negotiate the content of RMPs during the drug submission review process. In addition, stakeholders indicated that RMPs should not affect the practice of health care professionals (e.g. controlled distribution programs that would impact a pharmacist’s ability to dispense a drug).
HTA organizations indicated that they would like additional operational and implementation details on how RMPs would be reviewed by Health Canada because of an anticipated impact on their review processes.
Rolling reviews (amendments to FDR)
HTA organizations also wanted to better understand how rolling reviews would be implemented and the effect these proposed amendments might have on their review process. In addition, industry stakeholders expressed general support for the option of rolling reviews; however, they requested additional clarification on the types of drugs that would qualify for a rolling review and on how these amendments would be implemented. Stakeholders also communicated their expectations that the advantages of a rolling review should be evident compared to other priority submission options.
Modernizing requirements for biologic drugs (amendments to the FDR)
With regard to the modernization of provisions respecting biologics, stakeholders indicated that the final regulatory approach for lot release testing should be risk-based and flexible. Health Canada currently uses the risk-based tiered approach outlined in Guidance for Sponsors: Lot Release Program for Schedule D (Biologic) Drugs. The proposed regulatory amendments would allow for the continued use of a risk-based approach.
Stakeholders were in favour of modernizing labelling requirements for biologics but suggested that spacing considerations be made for small labels, that the acceptance of universal labels and machine-readable codes be considered, and that any changes, like the proposed requirement to indicate the species of origin, should not impact the ability of health care professionals to prescribe certain medications. Multiple stakeholders stated that if alternate means of indicating the expiry date on stockpiled drugs were to be allowed, the requirement should ensure that the date is quickly and readily available for professionals and patients using the drug. The proposed regulatory amendments for the labelling of biologics in small containers would provide manufacturers with flexibilities already provided in Division 1 for other drugs. At this time, there are no restrictions on the use of universal labels or machine-readable codes, as long as they meet the requirements under Canadian law. Health Canada continues to support the ability of health care professionals to prescribe medication, including by ensuring that information about a drug is clear and accessible.
Although OSEs are a current practice, some industry stakeholders commented that they are a Canadian-specific requirement and therefore represent an additional burden. Some stakeholders suggested that Health Canada eliminate this practice in favour of leveraging information from other sources, such as GMP inspections. Other stakeholders indicated that OSEs should be flexible and risk-based, or further aligned with product-specific inspections like those conducted by the EMA or USFDA. GMP inspections differ from OSEs in that GMP inspections are not product-specific. OSEs are a critical tool used by the Department in a risk-based manner for a subset of drug submissions. It is a submission-related activity, assessing the suitability of the manufacturing process for the product, validating the information submitted in the drug submission and evaluating the manufacturing process as implemented at the site. Health Canada currently applies a risk-based approach in deciding whether additional information obtained from OSEs regarding the implementation of the manufacturing process in the proposed facilities is required to support decision-making for new drug or supplemental new drug submissions for biologics. The risk-based approach considers factors such as the complexity or novelty of the manufacturing process, Health Canada’s familiarity with the site, and the experience that the manufacturer has in producing a safe and consistent drug. The proposed regulatory amendment would provide transparency regarding the inclusion of information obtained directly at, or indirectly from, manufacturing sites in the decision-making process for the market authorization.
Out of scope
Health Canada also received a number of comments that were out of the scope of this regulatory proposal. These included comments regarding the authorization process for clinical trials, flexibilities related to labelling and market access for generic drugs, rolling reviews for drug establishment licences and medical devices, and amendments to authorization fees for veterinary drugs in order to facilitate market access and compete globally.
Modern treaty obligations and Indigenous engagement and consultation
As required by the Cabinet Directive on the Federal Approach to Modern Treaty Implementation, a detailed assessment of modern treaty implications was conducted on the proposal. The assessment did not identify any modern treaty implications or obligations.
Instrument choice
Health Canada considered the following regulatory and non-regulatory options.
Option 1: To not introduce regulatory amendments and maintain status quo
Without the proposed amendments to the FDR and MDR, the Department would not meet its objective to provide clear regulatory authority for flexibilities, some of which are currently in place by way of policy, to keep pace with innovation and facilitate access to advanced treatments and promising therapies, while continuing to ensure authorized drugs and licensed medical devices are safe, effective, and subject to appropriate oversight, as per its commitment made in the Health and Biosciences Roadmap. The absence of amendments to codify these flexibilities results in a lack of certainty for manufacturers that the agilities would be consistently available and applied.
Furthermore, legally binding commitments for the management of risks and uncertainties by way of T&Cs would continue to apply to opioids and designated COVID-19 drugs only. Under the MDR, T&C powers would be limited to tests for Class II to IV devices, while broad T&Cs would only apply to COVID-19 medical devices authorized under the current interim order.
The option to retain the status quo does not ensure that there are legal foundations in place to implement a drug and device framework that effectively oversees regulated drugs and medical devices throughout their life cycle.
Option 2: Propose regulatory amendments to introduce modernization elements to the regulatory framework for drugs and medical devices
This is the preferred option as it would ensure that the Minister has the appropriate tools for oversight of the safety and effectiveness of drugs and medical devices through enforceable T&Cs. It would move Canada from a legal framework where Health Canada passively reacts to problems to one where risks are proactively mitigated through regulatory requirements such as T&Cs and RMPs. It would also support timely access for Canadians to critical drugs by including an option for a rolling review.
This option would allow the Department to begin to address the commitments made in the Health and Biosciences Roadmap, to reduce the burden on industry and to bring its regulations up to date with current practice so they are clear to stakeholders.
Regulatory analysis
Benefits and costs
The cost-benefit analysis (CBA) aims to quantify the benefits and costs of the proposed Regulations Amending Certain Regulations Made Under the Food and Drugs Act (Agile Licensing).
The full CBA report is available upon request.
Cost-benefit statement
The incremental costs to the industry are estimated at $158 million (PV) over a 10-year period. It is expected that the cost to Government over the same period will be $26 million (PV) to review and manage T&Cs and rolling reviews. As a result, the total anticipated costs of the proposed regulations in PV terms is $184 million over a 10-year period, discounted at 7%, or an annualized average of about $26 million.
The quantified benefits include an estimated $70 million in PV in sales from achieving market authorization two months earlier than under the status quo. Canadians should benefit from an improvement in their quality of life as a result of $302 million in PV. In addition, the elimination of regulatory requirements around standards should benefit industry by $68 million (PV). The total expected benefit in PV terms is $440 million over a 10-year period, discounted at 7%, or an annualized average of approximately $63 million.
- Number of years: 10 (2024 to 2033)
- Price level year: 2021
- PV base year: 2024
- Discount rate: 7%
| Impacted stakeholder | Description of benefit | Year 1 | Year 2 | Year 3 | Final year | Total (PV) | Annualized value |
|---|---|---|---|---|---|---|---|
| Industry | Two months of sales | $0 | $10,710,000 | $10,710,000 | $10,710,000 | $69,778,137 | $9,934,837 |
| Industry | Standards | $9,112,500 | $9,112,500 | $9,112,500 | $9,112,500 | $68,482,554 | $9,750,375 |
| Canadians | Two months of QALY table b1 note a | $0 | $44,720,676 | $45,167,883 | $48,426,084 | $301,944,819 | $42,990,149 |
| All stakeholders | Total benefits | $9,112,500 | $64,543,176 | $64,990,383 | $68,248,584 | $440,205,510 | $62,675,361 |
Table b1 note(s)
|
|||||||
| Impacted stakeholder | Description of cost | Year 1 | Year 2 | Year 3 | Final year | Total (PV) | Annualized value |
|---|---|---|---|---|---|---|---|
| Government | T&Cs | $0 | $3,145,964 | $3,145,964 | $3,145,964 | $20,496,684 | $2,918,267 |
| Government | Rolling reviews | $131,888 | $635,895 | $635,895 | $635,895 | $4,274,892 | $608,648 |
| Government | IT solutions | $1,028,207 | $65,887 | $65,887 | $65,887 | $1,457,476 | $207,512 |
| Industry | T&Cs — Drugs | $0 | $7,293,333 | $14,586,667 | $21,880,000 | $122,550,603 | $17,448,449 |
| Industry | T&Cs — Medical devices | $0 | $1,090,000 | $2,180,000 | $3,270,000 | $18,315,378 | $2,607,698 |
| Industry | RMPs | $0 | $2,289,600 | $2,289,600 | $2,289,600 | $14,917,276 | $2,123,884 |
| Industry | Rolling reviews | $0 | $360,000 | $360,000 | $360,000 | $2,345,484 | $333,944 |
| All stakeholders | Total costs | $1,160,095 | $14,880,679 | $23,264,012 | $31,647,346 | $184,357,792 | $26,248,402 |
| Impacts | Year 1 | Year 2 | Year 3 | Final year | Total (PV) | Annualized value |
|---|---|---|---|---|---|---|
| Total benefits | $9,112,500 | $64,543,176 | $64,990,383 | $68,248,584 | $440,205,510 | $62,675,361 |
| Total costs | $1,160,095 | $14,880,679 | $23,264,012 | $31,647,346 | $184,357,792 | $26,248,402 |
| NET IMPACT | $7,952,405 | $49,662,497 | $41,726,370 | $36,601,238 | $255,847,718 | $36,426,959 |
In terms of the qualitative benefits, the proposed amendments are expected to provide clarity to both industry and the regulator, allowing greater flexibilities for dealing with future innovations. Furthermore, they would contribute to the life cycle approach to the regulation of therapeutic products necessary to protect Canadians from harm from the therapeutic products they use while providing them with therapeutic benefits. The result of a life cycle approach should be a reduction in adverse reactions and medical device incidents.
Baseline scenario
The baseline reflects current business and review processes, and is used as the basis for the calculation of any incremental costs.
Prior to enacting Vanessa’s Law (2014), Health Canada had limited options when a serious safety issue arose concerning a marketed product. The Minister did have the ability to impose T&Cs with respect to tests on a medical device licence but not on any other product. Health Canada could also keep a product on the market while issuing a safety warning and possibly negotiating a label change with the manufacturer. Otherwise, the product would have been removed from the market, potentially depriving Canadians of life-saving marketed products. The life cycle powers introduced by Vanessa’s Law at the Act level improved the regulatory options to keep products on the market while improving the ability of Health Canada to address safety issues.
These authorities have helped move Health Canada towards a life cycle approach to product regulation; however, a number of Health Canada policies are still exercised through guidance.
Terms and conditions
The Minister currently has the authority to impose T&Cs related to testing on Class II to IV medical device licences and to amend T&Cs to take into account any new development with respect to the device. Currently, regulations also allow the Minister to impose T&Cs on the drug identification number for opioids and designated COVID-19 drugs. Under Health Canada’s NOC/c policy, manufacturers accept to adhere to conditions intended to confirm the effectiveness of new human drugs with promising clinical evidence to treat serious or life-threatening conditions. The conditions under the NOC/c policy are similar to T&Cs in other jurisdictions; however, they rely on voluntary compliance and are not enforceable. The NOC/c policy has not been applied to veterinary drugs nor have T&Cs been imposed on drug identification numbers for veterinary drugs.
Risk management plans
Currently, manufacturers voluntarily comply with guidance on the submission of RMPs. Typically, RMPs are submitted in the same format as required by the EMA. Health Canada assumes some costs from negotiation during the submission review process to bring RMP content in line with recommendations in guidance for Canadian-specific content. As RMPs are not a requirement in regulation, the Minister has limited authority to compel a manufacturer to submit a complete RMP in a timely manner, which leads to additional time being spent negotiating with the manufacturer.
Rolling reviews
Currently, the FDR explicitly contemplates rolling reviews for designated COVID-19 drugs. Rolling reviews are also currently available for veterinary drugs where the Minister has indicated the intent to conduct the review in collaboration with a foreign regulatory authority, and for annual updates to seasonal influenza vaccines.
Regulatory scenario
The regulatory amendments are described under each proposed element below.
Costs to industry
Terms and conditions for drugs
Under the proposed regulations, T&Cs could be imposed at any time, but it is intended that they would not be imposed on every authorized drug. They would be used to manage the uncertainties relating to benefits and/or risks (e.g. request for confirmatory clinical studies, quality and/or effectiveness studies).
As per criteria under the NOC/c policy, conditions are currently applied to new human drugs with promising clinical evidence to treat serious or life-threatening conditions. The incremental costs and benefits would be measured against the current practice of the NOC/c policy.
The NOC/c policy does not apply to veterinary drugs; therefore, to date, there have not been any NOC/c nor T&Cs applied to veterinary drugs. However, the proposed regulatory amendments defining T&Cs would allow manufacturers of veterinary drugs to be subject to this authority.
On an annual average, Health Canada issues seven NOC/c. Based on internal analysis and the expanding scope of the proposal, it is anticipated that there would be an increase of 10 T&Cs issued annually. Through a costing survey sent to industry (Agile CBA Survey), conducted between July and October 2021, stakeholders indicated a wide range of costs of T&Cs, both based on their experience with NOC/c and T&Cs in other jurisdictions, and anticipated costs. The reported costs range from $4,000 (submitting study results) to $6.2M (conducting a clinical study). Based on the responses from those who had experience with NOC/c and T&Cs, the average costs of T&Cs are estimated to be $2.2M. These costs would include providing clinical study reports for ongoing studies, monitoring and providing any updates on disease changes, and providing additional information.
It is assumed that T&Cs would resolve within three years, as indicated by some stakeholders, and that their cost would distribute equally over this period.
The estimated cost of T&Cs to the industry in the first year of implementation (second year of the regulation) is $7,293,333, the second year is $14,586,667 and every year after is $21,880,000. The PV is approximately $122,550,603 over a 10-year period, discounted at 7%.
Terms and conditions for medical devices
Under the current MDR, manufacturers are required to comply with T&Cs that are imposed on their Class II to IV medical device licences, and the Minister’s ability to impose T&Cs is limited to testing requirements. The proposed regulations would expand the scope of T&Cs beyond testing requirements, where T&Cs can be imposed or amended at licensing or after licensing and are expected to pertain mainly to uncertainties relating to the benefits and/or risks of a medical device.
It is expected that T&Cs could be used to assess the long-term safety or effectiveness of a medical device in response to evidence, including real-world evidence, not being available at the time of licence issuance. They could also be used to collect specific post-licence data for underrepresented subsets of clinical evidence to enhance labelling or statistics for specific subpopulations (e.g. pregnant women, pediatrics).
On average, annually, there are approximately 1 810 medical device licences authorized and 150 T&Cs issued. Under the proposed regulations, it is anticipated that there would be an increase of 5% to 10% of T&Cs issued. To be conservative, it is assumed T&Cs would increase by 10% or 15 additional T&Cs would be issued per year. Moreover, historical data has shown that T&Cs are typically resolved within three years. Similar to the treatment of T&Cs for drugs, the cost of complying with T&Cs for medical devices would be distributed over the three-year period.
Based on the responses to the costing survey, the costs of fulfilling T&Cs range from $4,000 to $1M. This is based on stakeholders’ experience with T&Cs, including but not limited to their activities ranging from submitting final study results, additional non-clinical data, and/or study results of an ongoing study to a new clinical study to confirm effectiveness. The average cost of fulfilling T&Cs is $218,000.
The expected cost of T&Cs for medical devices in the first year of implementation (second year of the regulation) is $1,090,000, the second year is $2,180,000 and every year after is $3,270,000. This translates to a PV of $18,315,378 over a 10-year period, discounted at 7%.
Risk management plans
The proposed regulations would formalize the practice of requesting RMPs from manufacturers of human drugs. As per current practice, manufacturers would be required to include Canadian-specific information as applicable to the Canadian context and marketplace. The Minister could also require an updated RMP if there were reasonable grounds to believe that the risks and uncertainties associated with the drug are significantly different from the existing plan or if the drug presents a serious risk of injury to human health that warrants measures to reduce the probability or severity of such an injury that are significantly different than those described in the existing plan. The amendments would also create a new obligation for the manufacturers to submit a summary of new and updated RMPs in both official languages.
Currently, Health Canada receives an average of 103 new RMPs annually as per Guidance. All of these new RMPs are voluntarily submitted upon request. Of the 103 RMPs, most already have summaries provided but only in English; therefore, it is expected manufacturers would have to translate these RMP summaries. In addition, Health Canada receives an annual average of 321 updated RMPs. Under the proposed regulations, all 321 updated RMPs would require a summary in both languages.
In response to the Agile CBA Survey, stakeholders provided a wide range of costs from developing a core RMP, to creating a summary, to translating. It should be noted that most of the costs provided are based on actual experience of developing RMPs either in Canada or in other jurisdictions. It is recognized that translating an updated summary would require significantly less time and effort than preparing the initial summary, as most changes to a summary involve adding or removing a safety concern or a risk minimization measure; therefore, the cost of translating an updated summary should also be less. However, due to the lack of data and also to be conservative, Health Canada is using the cost to translate a new summary as a proxy for the cost to translate an updated summary.
It is anticipated that a total of 424 (103 + 321) summaries will be translated per year at a cost of $5,400 per summary. This equates $2,289,600 annually, non-discounted. The PV is $14,917,276 over a 10-year period, discounted at 7%.
Rolling reviews
The proposed amendments would allow for rolling reviews in order to facilitate timely access to human and veterinary drugs that are needed to address emerging infectious diseases in Canada, and drugs for the treatment, prevention or diagnosis of serious or severely debilitating diseases or conditions.
The proposed pre-submission requirements include a rolling review application package that would include information demonstrating how the drug to which the submission would relate meets the eligibility criteria. This is similar to the clinical assessment package provided under the current priority review policy. In addition, prior to filing the submission, the manufacturer would be required to provide the Minister with a submission plan that would set out how and when the manufacturer intends to provide any missing information. It is expected that the missing information would be submitted in two to four transactions within the time frame set out in the notice. Stakeholders indicated the estimated cost to develop and submit a submission plan is between $40,000 and $59,000. The cost is varied due to whether a submission plan is a replica from a global rolling review submission or is prepared for Canada only. The average cost of a submission plan is assumed to be $49,500. Also, as indicated by stakeholders, the cost associated with providing any missing information is approximately $7,500 per transaction. It is assumed that there would be five rolling reviews of human drugs per year and the missing information would be provided on average over three transactions per submission.
The rolling review is an option for certain drugs, since manufacturers can instead wait and file a non-rolling submission or rely on expedited review policies such as priority review. Therefore, the total estimated additional cost to use the rolling review pathway rather than other review options for five submissions per year is $360,000. The anticipated cost in PV is $2,345,484 over a 10-year period, discounted at 7%.
Rolling reviews are currently in practice for annual updates to seasonal influenza vaccines and for veterinary drugs where the Minister has indicated the intent to conduct the review with a foreign regulatory authority. The amendments would provide a regulatory framework for these existing practices. In the context of Health Canada, joint or parallel reviews are conducted with international regulators, most notably the USFDA’s text-center for Veterinary Medicine under the Regulatory Cooperation Council.
It is not anticipated there would be incremental costs for rolling reviews for annual updates to seasonal influenza vaccines or for veterinary drugs where the Minister has indicated their intent to conduct the review with a foreign regulatory authority, as the proposed regulations are designed to codify what has been in practice.
Pre-positioning
It is not anticipated that there would be any impacts to industry, as the proposed amendments would continue to limit pre-positioning to drugs akin to those that treat COVID-19, namely public health emergency drugs. The decision to consider pre-positioning of a public health emergency drug would continue to be driven by the Chief Public Health Officer of Canada for conditions set out on the List of Conditions that Threaten Public Health.
Assuring drug quality during manufacturing
The proposed amendments to Part C, Division 2, are not anticipated to introduce any additional cost or burden on industry. The amendments would support the intent of existing quality control requirements by clarifying interpretations for GMP compliance that are set out in Health Canada’s guidance documents Good manufacturing practices guide for drug products (GUI-0001) and Guidelines for environmental control of drugs during storage and transportation (GUI-0069).
Modernizing requirements for biologics
It is not anticipated that there would be any incremental costs to industry, as the proposed amendment would repeal outdated product-specific provisions spanning between C.04.050 and C.04.683, and other elements of the amendments would reflect current practice as well.
Information considered to support the examination of drug submissions
It is not anticipated that there would be any incremental costs to industry, as the proposed regulations are in line with current practice.
Disaggregated clinical trial data for new human drug submissions and supplemental new human drug submissions
It is not anticipated that there would be any incremental costs to industry, as manufacturers would only be required to submit clinical trial data broken down into population subgroups to support the safety and effectiveness of the new (or supplemental) drug submission if the disaggregated data has already been submitted to the USFDA or the EMA.
Standards
Current regulations require that the inner and outer labels of certain drugs indicate if a prescribed, Schedule B compendium or manufacturer’s standard has been used for a drug. This requirement applies to drugs regulated solely under Part C, Division 1, and those regulated under Division 8 that are not also regulated under Divisions 3 and 4 (i.e. radiopharmaceutical and biologic drugs). Health Canada is proposing to remove this labelling requirement. It is not expected that this element of the proposal would impose any incremental costs to industry. Removing this labelling requirement would allow manufacturers the space to provide information that is more relevant to health care providers and patients.
Presently, if a manufacturer’s standard is used, the regulations require that the drug meet the highest degree of purity and the least variation in potency of any pharmacopoeia in which it is listed under Schedule B of the Act. This applies to drugs regulated solely under Division 1 and those regulated under Division 8 (including those that are also regulated under Divisions 3 and 4 [i.e. radiopharmaceutical and biologic drugs]). It is proposed that drugs regulated under Division 8 (other than those listed in Schedule C to the Act) in respect of which a manufacturer’s standard is claimed would be exempt from this regulation (e.g. manufacturers would be allowed to qualify limits for purity and/or potency beyond those listed in a Schedule B pharmacopoeia that would be acceptable to Health Canada). It is not expected that this element of the proposal would impose any incremental costs to industry, and it would reduce unnecessary regulatory burden.
Costs to Government
Terms and conditions for drugs and medical devices
Uncertainty exists over the frequency and type of T&Cs that Health Canada may issue in the future. There are two factors which could create an increase in the number of T&Cs issued. The first is an increase in receipt of applications for drugs in areas of unmet need such as oncology or pediatric rare diseases (where the likelihood of applying T&Cs is relatively high). The second is a general increase in use once the broader T&C authorities for drugs and medical devices come into force, particularly where risks or uncertainties might not have been identified at the time of authorization. The CBA recognizes an initial incremental increase in the number of T&Cs issued due to the proposed regulations; however, it is expected there would be no additional growth over the analysis period because many factors (such as feasibility, less burdensome means, or application of the existing requirements) would be taken into account before imposing T&Cs.
The cost to government for the management of T&Cs is highly variable and is dependent upon the risk of the assessed product and the requirements stipulated in the T&Cs. The cost of managing these T&Cs ranges from $119,994 to $167,982, or on average $143,988, per T&C imposed at the time of issuance of the market authorization. The cost of managing T&Cs issued post-authorization ranges from $159,568 to $180,562 or on average, $170,064 per T&C. It is anticipated that 10 T&Cs for drugs (5 at the time of authorization and 5 post-authorization) would be managed annually by Health Canada, representing an annual cost of $1,570,266 or $10,230,649 PV over a 10-year period, discounted at 7%.
There are approximately 150 medical devices issued with T&Cs attached under the MDR, and it is estimated that this would increase by 5–10% annually due to the proposed expanded scope of T&Cs. If the upper bound were used, there would be approximately 15 new T&Cs issued annually at an average cost of $105,046, resulting in management costs of approximately $1,575,698 per year or $10,266,035 PV over a 10-year period, discounted at 7%.
Additional costs to Government may also be incurred where compliance verification or enforcement actions are undertaken respecting T&Cs. Health Canada’s choice of a particular compliance and enforcement action is informed by a risk-based approach and would depend on the type of T&C. Enforcement actions are expected to be rare and reserved for cases where it is necessary to address a known or suspected violation of T&Cs.
The total cost to Government for the management of T&Cs for drugs and medical devices is anticipated to be $3,145,964 annually or $20,496,684 PV over a 10-year period, discounted at 7%.
Risk management plans
Health Canada currently receives RMPs voluntarily; however, the Government often negotiates with applicants to bring the RMP contents in line with what is described in guidance, particularly for inclusion of an addendum covering Canadian-specific information. At present, comments and recommendations that are provided by Health Canada are addressed approximately 90% of the time, with 70% of these submissions being “accepted with minor changes.” Almost all manufacturers include a summary with their RMP submission and approximately 40% include an addendum of Canadian-specific information.
Incremental costs for Government may arise from compliance and enforcement activities that would be necessary to address a failure by an organization to submit an updated plan in accordance with the proposed regulations; however, these cases are expected to be rare.
Rolling reviews
The proposed amendments would make the option of a rolling review available for both new drug submissions and supplemental new drug submissions for certain drugs. While the amendments would allow a manufacturer to provide components of the submission in sequential transactions as information becomes available after the drug submission has been filed, the threshold and standard of data required to obtain an authorization would be the same as for a submission reviewed without a rolling review. Although the information required for a rolling review would be exactly the same as for a non-rolling new drug submission or supplemental new drug submission, the review process would be expected to be more resource intensive for Health Canada due to the incremental approach to review.
Each new transaction received in accordance with the rolling review option would be expected to require additional time for
- Revisions of RMPs (once enacted in regulations);
- Increased interactions during the pre-market phase;
- Sponsor communications (i.e. the review of the submission would resume at disparate times as information is provided for review);
- Review of educational materials;
- Additional labelling input; and
- Additional review time for approval in the final review stage.
New one-time costs would also arise from the necessary revisions to standard operating procedure documents (SOPs) to include updates to training, development of processes and process materials, and new oversight. This is estimated at $131,888 in the first year.
All incremental costs are assumed to be due to non-contiguous review of the information provided to support the submission, as well as the processing of missing information after the submission that would have been filed.
While the content of a submission for rolling review would be the same as for the equivalent submission without a rolling review, due to the periodic nature of the review and consistency required over the entirety of the review process (preferably by the same review team) each submission is estimated to cost Health Canada an additional $127,179. Assuming there would be 5 submissions, the anticipated total cost in subsequent years is $635,895 or $4,274,892 PV over a 10-year period, discounted at 7%.
Also, there is an anticipated one-time cost of $1,028,207 to account for an update to the information technology (IT) infrastructure. This update would be necessary to handle the extra transactions for rolling reviews, T&Cs and RMPs. It would also be to account for docuBridge (a submission management system) updates, application form changes, training and implementation. For each subsequent year, there would also be a cost of $65,887 to manage the additional transactions for rolling reviews, T&Cs and RMPs. This translates to a total cost of $1,457,476 PV for IT solutions over a 10-year period, discounted at 7%.
Pre-positioning
The proposed amendments would allow a drug to be pre-positioned not only in response to the COVID-19 pandemic but for other public health emergencies that would be referred to on the List of Conditions that Threaten Public Health. The proposed amendments are not anticipated to result in any new or additional information to be submitted or reviewed and would not impact current government operations. Anticipated costs are expected to be negligible.
Assuring drug quality during manufacturing
No changes are anticipated to Health Canada’s oversight of drugs or current operational practice with respect to GMP inspections as a result of the proposed amendments. No additional costs to Government are anticipated.
Modernizing requirements for biologics
The amendments are intended to bring current operational and industry practice into regulations. Given that the baseline of the cost analysis is current practice, no additional costs to Government are anticipated as a result of these amendments.
Information considered to support the examination of drug submissions
No costs to Government have been identified.
Disaggregated clinical trial data for new human drug submissions and supplemental new human drug submissions
It is not anticipated there would be an impact on the number of submissions as a result of this regulatory amendment. The proposed requirement could increase the quality of the submitted data, as reviewers already analyze any disaggregated data voluntarily provided, and this would continue to be reviewed as they do currently. There could be an additional screening step added, but the additional work is not expected to be significant and would be managed by existing staff. Reviewer training would be required as well as the development of SOPs, but resources required are part of the implementation of the Health Products and Food Branch’s Sex- and Gender-based Analysis Plus Action Plan.
Standards
No costs to Government have been identified.
Benefits
The proposed amendments would modernize the language in the FDR and MDR, providing clarity to both industry and the regulator allowing greater flexibility for dealing with future innovations. While it is acknowledged that many of the provisions proposed in these amendments simply codify existing practice, the clarity provided is expected to promote greater innovation and earlier access to products in Canada and provide some efficiencies to industry, payers, and medical professionals while improving safety and effectiveness, and resolving uncertainty.
Combined, the proposed regulations would contribute to the life cycle approach to the regulation of therapeutic products necessary to help Health Canada deal with the balance between protecting Canadians from harm from the therapeutic products they use and providing them with therapeutic benefits.
Benefits to industry
Terms and conditions for drugs
The cost of developing a drug to market has been estimated to be between $800 million and $2.5 billion when all research and development costs, including drug failures, are included. The process takes between 9 and 15 years from discovery to approval.footnote 3 During the drug development process, approximately 60% of all drugs fail due to the lack of evidence of safety and effectiveness. Once a drug is on market, its performance in real-world conditions can be different from that in a clinical setting. A major failure in safety or effectiveness not only puts the health and safety of patients at risk, but the subsequent removal of a drug can cost industry millions of dollars. In some cases, the drug may have been very beneficial to a segment of the population while only posing a risk to another segment. The proposed regulatory amendments are meant to clarify existing provisions, to provide faster or more flexible market access, to prevent shortages, and to help manage uncertainties and risks to reduce the probability of product failure.
The application of T&Cs imposed on a drug identification number, either at the time of authorization or post-authorization, alongside the use of RMPs would help identify safety and effectiveness issues sooner, allowing Health Canada to apply risk mitigation strategies to address real world safety signals to reduce harm, while allowing a product to continue on market with appropriate safety oversights where deemed necessary. This would potentially allow industry to keep a product on market without disruptions.
Terms and conditions for medical devices
The amendments would enhance Health Canada’s capacity for continued oversight, assessment, and communication at both the licensing stage and once medical devices are on the market. For industry, this could mean the rapid identification and resolution/mitigation of risks that arise while still allowing devices to remain on the market without disruption.
Risk management plans
No benefits to industry have been identified.
Rolling reviews
For rolling review of drugs such as the annual influenza vaccines, the proposed amendments serve to codify existing practice and represent no benefits, as industry is already operating as if the provisions were in place.
In the case of a major event similar to the COVID-19 pandemic, the Regulations would provide industry with clear requirements for a rolling review to address a public health emergency added to a list, which would be incorporated by reference.
For those drugs that meet the eligibility requirements to access the rolling review option, earlier market access of up to six months could be possible compared to the standard process. Based on a review of Health Canada’s priority review timelines and the types of drugs expected to qualify for a rolling review, it is estimated that these drugs would be approved on average two months earlier than under current processes. While there may be some costs associated with being on market an additional two months, the majority of costs involved are assumed to be carried in getting to market; therefore the value of sales is used as the measure for the benefit. In 2019, prior to the pandemic, sales of patented medicines in Canada reached $17.2 billion on 1 364 patented medicines, giving an average annual revenue of $12.6 million per patented drug.footnote 4 The average of the top 10 drug sales in Canada for 2020 was over $500 million with the highest sales being in excess of $1 billion.footnote 5
Assuming five drugs with average annual sales ($12,600,000) being approved two months (17%) earlier, the rolling review option could provide a benefit to industry of $10.7 million in sales.
5 drugs × $12,600,000 × 17% = $10,710,000
The anticipated quantifiable benefit to industry is $10,710,000 annually or $69,778,137 PV over a 10-year period.
Assuring drug quality during manufacturing
The expected benefit is clarity around the Regulations.
Modernizing requirements for biologics
Since the regulatory amendments serve to modernize the Regulations and would reflect current practice, the anticipated benefit is greater clarity to industry.
Information considered to support the examination of drug submissions
The expected benefit is clarity around the regulatory intent.
Disaggregated clinical trial data for new human drug submissions and supplemental new human drug submissions
No benefits to industry have been identified.
Standards
Manufacturers would no longer need to change their label if it became necessary for them to change the standard of their drug. The proposal would open up some space on the label, therefore allowing manufacturers the flexibility to provide other valuable information to patients and health care practitioners. Moreover, for drugs that claim a manufacturer’s standard, the amendments could reduce the time manufacturers are required to spend monitoring the pharmacopoeia and the cost of retesting drugs to meet the strictest standards.
While the benefit is not limited to only generic drug manufacturers, they would be most likely to benefit from the proposed regulatory change. Stakeholders indicated savings from labelling ranging from $120,000 to $400,000 annually; this would include disposal cost for labels. Furthermore, savings from manufacturer’s standards could range from $45,000 to $650,000 a year in respect of monitoring and retesting drugs. The administrative cost savings of monitoring Schedule B pharmacopoeias, assessing the impacts and reporting to Health Canada is estimated to be about 37.5 hours for each pharmacopoeia and assumes there are three publications per year. Taking an average approach, annual cost savings from labelling and manufacturer’s standards would be about $260,000 and $347,500, respectively. Given that there are 15 firms supplying generic products in Canada, it is estimated that the savings from the proposed regulations could be $9,112,500 per year.
15 firms × ($260,000 + $347,500) = $9,112,500
It is expected that the proposed amendment would yield annual savings of $9,112,500 or $68,482,554 PV over a 10-year period. Furthermore, stakeholders also indicated that current regulations occasionally cause shortages, as the new standards need to be met. The proposed regulations would reduce such disruptions in sales.
Benefits to Government
Terms and conditions for drugs
The amendments would codify in law existing practices under the NOC/c policy and allow the imposition of T&Cs at issuance of market authorization and post-authorization. They could reduce the time required to achieve a resolution in discussions with a manufacturer around the manner in which to deal with the potential safety and uncertainty issues of a product. The amendments could also allow Health Canada to adapt to innovation and change.
Terms and conditions for medical devices
The amendments would codify in law existing T&Cs practices at Health Canada and broaden the scope of T&Cs that can be imposed on a medical device licence. The amendments could also allow Health Canada to adapt to innovation and change.
Risk management plans
Health Canada would be able to compel manufacturers to submit and amend RMPs to address any deficiencies in a timely manner where they may not do so voluntarily.
Rolling reviews
In the last 20 years, Canada has experienced three major disruptions in the form of significant infectious diseases: SARS in 2003, H1N1 in 2009, and COVID-19 in 2020. Drugs used to treat these types of communicable disease outbreaks could be approved using the rolling review option.footnote 6,footnote 7,footnote 8 Amendments to the regulations would likely allow for the rolling review option for drugs used to fight these types of pandemics, without having to use interim orders.
The use of rolling reviews for the seasonal flu vaccines is a well-established standard practice and no additional benefit is identified.
Modernizing requirements for biologics
Clearly written, modern regulations may reduce the number of enquiries from industry regarding the provisions, saving some time and resources for Health Canada.
Information considered to support the examination of drug submissions
These proposed amendments would provide clarity regarding the Minister’s authority to consider, as part of a submission, information obtained directly at, or indirectly from, a building or site where a drug is fabricated, packaged, labelled or tested, when making a decision about whether to authorize the drug.
Disaggregated clinical trial data for new human drug submissions and supplemental new human drug submissions
The proposed requirement could increase the utility of the submitted data, making identifying any potential differences in safety or efficacy between subpopulations more apparent.
Standards
No benefits to government have been identified.
Benefits to Canadians
Combined benefits
The use of T&Cs for drugs and medical devices and RMPs for drugs, as well as the receipt of disaggregated data, would increase the safety of these therapeutic products in Canada, and could help to reduce the incidence of adverse drug reactions (ADRs) and medical device incidents (MDIs), and manage uncertainties around safety and effectiveness of drugs and devices under real-world conditions. When used in conjunction with a rolling review, there is the potential for new and urgently needed drugs to achieve market authorization sooner.
Terms and conditions for drugs
The ability to apply T&Cs at the time of authorization is currently limited to designated COVID-19 drugs and certain opioids. Under the proposed regulations, their expanded use to all drugs at the time of authorization, as well as in the post-authorization phase, should improve product safety.
Despite rigorous evaluations, monitoring of real world safety and effectiveness often identifies harms and uncertainties not demonstrated during pre-market clinical evaluations. In 2019, Health Canada received over 96 000 domestic ADR reportsfootnote 9 relating to drugs and it has been estimated that only 10% of all ADRs are reported.footnote 10 Estimates show that as many as 1 in 1 000 Canadians under the age of 65 will go to hospital as a result of an ADR in their lifetime. The number jumps to 1 in 200 for Canadians over the age of 65.footnote 11 Of over 96 000 ADRs reported to Health Canada, 18 852 reports mentioned hospitalization; ADRs may contribute to as many as 6 119 deaths.footnote 9 Women are also more likely to experience an ADR than men.footnote 12 Pediatric medicines rarely have the same information available as drugs for adults given the obvious ethical limitations on testing products on children. Other populations who are generally under-represented in the clinical trials conducted in preparation of a regulatory drug submission are similarly likely to see benefits.
The direct medical cost of ADRs has been calculated to be as high as $20B (in 2019 dollars) annually.footnote 13 Based on the data reported to Health Canada in 2019, 19.5% of ADRs were linked to hospitalization and 2.6% reported of a potential life-threatening condition.footnote 14 While it is not possible to determine to what extent the proposed regulatory amendments will reduce the frequency of ADRs in Canada, the potential impact could be significant. For example, even a 0.1% decrease in ADRs would reduce the number of deaths by 6 (6,119 × 0.1%), which when applied to the $8.59M value of a statistical life used in Canada,footnote 15 would represent a benefit of $50M. Similarly, a 0.1% decrease in the estimated direct medical annual cost of $20B would represent savings of $20M per year. However, it is not possible to estimate the reduction in ADRs that might result from this regulation.
In addition to the direct cost of an ADR to the health care system or an individual, family members and other caregivers must take time off to care for a senior, child, or other persons that have been hospitalized or otherwise incapacitated. A reduction in the number of ADRs as a result of improved life cycle oversight would reduce the hardship of having to care for another. ADRs also cause absenteeism from work and presenteeism (working while sick with reduced productivity), affecting economic activity.
Terms and conditions for medical devices
The expansion of T&Cs would specifically address uncertainties and risks that only become apparent under real-world use of a device. This could be helpful in identifying the patient populations that would benefit the most from the mitigation of serious health risks. Applying T&Cs may reduce MDIs and prevent the withdrawal of a product from market that could have been used safely with the appropriate risk mitigation strategies.
Risk management plans
No benefits to Canadians have been identified.
Rolling reviews
Canadians are expected to benefit from rolling reviews for the approval of eligible human and veterinary drugs. Under the status quo, all of the information required for the Minister to assess the safety and effectiveness of a new drug is available for review at the time of submission. The regulatory amendment could allow specific drugs and vaccines to reach the conclusion of Health Canada’s review process up to 6 months earlier as the review will begin before the full package of data is available (the length of time is dependent on whether it is measured against a standard review or against priority review). This will allow earlier approval, which could lead to earlier access. This has the potential to provide significant benefits through the reduction in health care costs and improvements in quality of life across the population at large.
Health Canada, based on its experience with priority review, estimates that the creation of a new rolling review pathway will lead to five drugs per year being granted market authorization an assumed two months earlier. Depending on the patient population and type of disease, the benefits will vary widely. To assess the benefits of earlier access, assumptions must be made around the size of the prescribed patient pool, and the impact the prescribed drug will have on the quality of life of those taking it. To be conservative in its estimate, Health Canada assumes that drugs target small patient populations, such as those found in drugs for rare diseases. Patient populations could in fact be much larger in the case of oncological drugs or vaccines using the rolling option.
A rare disease is defined as one that affects no more than 5 in 10 000 Canadians, or a maximum of 17 750 patients. It has been estimated that 1 in 12 Canadians (2.7 million) are affected by a rare disease and that there are over 7 000 rare diseases in the world. This creates an average patient population size of 385 individuals, which, we assume, is the average minimum population size for any drugs approved through this pathway. Drugs in general are only effective in 60% of the population; therefore, the number of patients benefiting from a drug can be further reduced to 231. Since the review path is intended for all types of drugs that meet the requirements, this is assumed to create a floor for the number of patients treated.
2 700 000 individuals ÷ 7 000 rare diseases = 385 patients × 60% = 231 treated patients
Canada’s population between 2015 and 2020 had an average growth rate of 1%. Therefore, the number of treated patients should also increase by 1% per year. So, by the 10th year of analysis, the treated patient size should reach 250.
In Canada, the value of a statistical life (VSL) for use in economic analysis is currently set by Treasury Board at $6.4 million in 2007 dollars. Adjusting for inflation, the VSL in 2021 dollars would be $8.59 million. Using the average life expectancy in Canada (82 years),footnote 16 the average age of a Canadian (41 years),footnote 17 and a 7% discount rate, this gives a value of a statistical life year (VSLY) of $599,370 in 2021 dollars for the average Canadianfootnote 18 using the formula
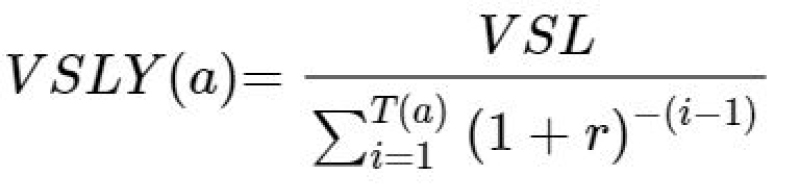
Text version: Formula to calculate the value of a statistical life year
VSLY at a is equal to the quotient of VSL over the sum of, open bracket 1 plus r close bracket, negative exponent open bracket i minus 1 close bracket, indexed from 1 to T open bracket a close bracket.
Where VSLY is the value of a life year, VSL is the value of a statistical life, r is the discount rate, a is the age, and i is the number of years remaining in life expectancy from age a.
The proposed regulations are assumed to allow for a minimum of 2 months of earlier access (0.17 years). While this could be life-saving in some instances (such as COVID-19 vaccination), it is more likely that this access will improve the quality of life for patients receiving treatment. The quality-adjusted life year (QALY) is a concept used in health economics to account for the fact that all years of life lived may not be equal due to illness experience across the life span. The QALY of full health is denoted as being equal to 1, and death as being equal to 0. Values for other disease states have been calculated to lie between these two ends. At any given point in life, the value of one year of life for a healthy individual is equal to the VSLY at that same time point. Various studies measuring the QALY of patients suffering from various diseases and conditions have generated a wide range of numbers. For example, the QALY for multidrug-resistant HIV was calculated to be 59% of that of a healthy person. Via survey, patients with treatable rare diseases and their caregivers reported having a QALY of 56% in the United States and 50% in the United Kingdom, while their estimate was 42% in the United States and 40% in the United Kingdom if the disease was untreatable.footnote 19 A recent study reviewed 494 new molecular entities approved by the FDA between 1999 and 2015 and found the average increase in the QALY of patients was 0.38.footnote 20 A 0.38 QALY improvement for an average-aged Canadian expected to live to 82 would represent a gain in VSLY of $227,760.
$599,370 VSLY × 0.38 QALY = $227,760 VSLYadj
Therefore, assuming an average of 5 drugs are approved each year, successfully treating 231 patients in year 1 for an additional 2 months (0.17 years) with an increased QALY of 0.38, the first-year benefit would be approximately $44.7 million. Assuming population growth, by year 10 the value would reach $48.4 million.
Year 2: 5 drugs × 231 patients × 0.17 years × $227,760 VSLYadj = $44,720,676
Year 10: 5 drugs × 250 patients × 0.17 years × $227,760 VSLYadj = $48,426,084
The total PV benefit over 10 years would be $301,944,819.
This number does not include the financial savings to family and other caregivers who spend time and resources caring for Canadians suffering from diseases, providing them with the supportive aids to daily living (e.g. personal care, meal preparation, cleaning, transportation, etc.) they require.
Pre-positioning
Canadians are expected to benefit from the proposed amendments. In the case of a future public health emergency, pre-positioning is expected to provide more timely access to drugs specific to the emergency. By amending the regulations to require that a licensed importer be identified to import the drug, health and safety risks would be mitigated through application of regulatory requirements that are appropriate to support the oversight of the activity of importation.
Modernizing requirements for biologics
No benefits to Canadians are expected as innovation will continue as currently takes place under guidance and current practice.
Information considered to support the examination of drug submissions
No benefits to Canadians are expected as this proposed amendment reflects current practice.
Disaggregated clinical trial data for new human drug submissions and supplemental new human drug submissions
While no direct benefits to Canadians are expected as a result of the proposed regulations, the requirement to provide disaggregated data may provide additional information respecting specific subpopulations that could lead to increased drug safety.
Standards
Industry indicated that the current requirement occasionally leads to drug shortages. Canadians would benefit from a more stable drug supply.
Small business lens
The small business lens would apply as all pharmaceutical, medical device and veterinary industry stakeholders would be impacted by the proposed regulations.
Using internal data, Health Canada estimated that approximately 26% of all impacted applicants or manufacturers would meet the definition of small businesses in Canada. The largest proportion of small businesses within their product category are medical device (36%) and veterinary drug (27%).
Although there would be no specific exemptions or processes proposed in the new regulations for small businesses, outside of what already currently exists in regulation and in practice, the needs of small business were taken into consideration in the creation of the regulations. Small businesses would also benefit from the greater flexibilities and clarity provided by the proposed amendment. For example, small pharmaceutical manufacturers could benefit from the flexibility provided for by earlier drug approval under the rolling review pathway, or spend their resources more efficiently as the proposed amendments would provide more clarity. Generally, many provisions would take any potential compliance and administrative costs, and/or adverse event consequences, into consideration prior to imposing T&Cs or an RMP on an applicant or manufacturer.
- Number of small businesses impacted: 225
- Number of years: 10 (2024 to 2033)
- Base year of costing: 2021
- PV base year: 2024
- Discount rate: 7%
| Activity | Annualized value | Present value |
|---|---|---|
| T&Cs | $4,471,413 | $31,405,337 |
| RMPs | $169,330 | $1,189,304 |
| Rolling reviews | $11,030 | $77,470 |
| Total compliance cost | $4,651,774 | $32,672,112 |
| Cost per impacted small business | $20,675 | $145,209 |
One-for-one rule
The one-for-one rule applies since there would be an incremental decrease in the administrative burden on business, and the proposal would be considered burden out under the rule. The proposed amendment to the requirements related to the standards would save drug manufacturers the time spent on monitoring Schedule B pharmacopeias and assessing the impacts. The resources required by businesses to monitor, assess the impacts and notify Health Canada when the standards have changed would be considered administrative burden. These activities require manufacturers to spend an average of 37.5 hours for each pharmacopeia publication at a rate of $31 an hour,footnote 21 and it is assumed there are three publications per year. The annual anticipated reduction in the administrative burden on businesses is estimated at $25,177 (in 2012 dollars) or $1,678 (in 2012 dollars) per business.
The objective of imposing T&Cs would be to manage the uncertainties relating to the benefits and/or risks of a product at the time of authorization or to manage emerging risks or uncertainties post-authorization. This requirement would be directly related to ensuring the health and safety of Canadians. Therefore, the costs of fulfilling the T&Cs are not considered as administrative burden as defined by the Red Tape Reduction Act as their primary purpose is not for ensuring compliance.
Currently, manufacturers are voluntarily submitting RMPs upon request as well as RMP summaries if available. Thus, it is not expected there would be incremental administrative requirements from the proposed regulations with respect to RMPs.
Regulatory cooperation and alignment
While these proposed regulatory amendments are not part of a formal regulatory cooperation plan with any foreign regulator, they would further align Health Canada’s requirements with those of other jurisdictions such as the United States, the European Union and Australia.
United States
The USFDA has the ability to compel, by law, an authorization holder to conduct tailored activities to manage risks and ensure the benefits of their drugs once on market, similar to the proposed amendments that would permit the Minister to impose T&Cs on any drug.
The USFDA has the ability to impose post-approval requirements on certain classes of medical devices. These conditions may be related to sale restrictions, periodic reporting, labelling requirements or testing, similar to the proposed amendments regarding T&Cs.
For certain drugs, the USFDA can compel, by law, submission of a plan outlining the manufacturers Risk Evaluation and Mitigation Strategies (REMS). REMS are similar to RMPs; therefore, these proposed amendments to the FDR would better align Canadian requirements for risk mitigation strategies with those of the USFDA.
The USFDA allows the use of rolling reviews for certain drugs provided they meet the criteria outlined under its Fast Track, Breakthrough Therapy, Accelerated Approval, Priority Review programs. Inclusion criteria include a drug that could be used to address a public health emergency, or a drug with preliminary clinical evidence indicating it may be a substantial improvement over existing therapies for serious conditions. Manufacturers that file submissions for rolling review with the USFDA experience more frequent pre-market interactions with the regulator and benefit from earlier market access, which would be the case for manufacturers that qualify for rolling reviews with Health Canada under the proposed amendments to the FDR.
Before the approval of a new drug application for some small-molecule pharmaceutical and biologic drugs, the USFDA evaluates the establishments through the use of on-site pre-approval inspections and/or by establishment file reviews. These are conducted by inspectors, investigators and other product specialists in a risk-based manner, accounting for facility risk, product risk and process risk. The inspection is performed to contribute to USFDA’s assurance that a manufacturing establishment named in a drug application is ready and capable of manufacturing a drug, and that submitted data are complete, accurate and consistent with that at the site of manufacture. This is analogous to Health Canada’s practice of conducting OSEs and the proposed amendment which would allow for the materials or information collected to support the assessment of a new drug submission.
The USFDA requires manufacturers to submit safety and effectiveness data by certain subpopulations (e.g. age, sex, race/ethnicity) and, in the case of effectiveness data, manufacturers must identify any modifications of dose or dose interval needed for these specific subpopulations.
The USFDA requires that a drug must meet the standard of the United States Pharmacopeia/National Formulary (USP/NF) if the drug appears in that pharmacopoeia, unless certain conditions are met. They do not require that a list of pharmacopoeia be monitored. Through these proposed amendments, manufacturers that claim a manufacturer’s standard for their drug authorized in Canada would also no longer need to monitor a list of pharmacopoeia, and update their product, if necessary. The USFDA does not require that a manufacturer include the standard of a drug on their label. The proposed amendments would be in alignment with this practice.
European Union
The European Union’s EMA has the ability to compel, by law, an authorization holder to conduct tailored activities to manage risks and ensure the benefits of their drugs once on market, similar to the proposed amendments that would permit the Minister to impose T&Cs on all drugs. The EMA may also set conditions on medical devices in exceptional cases relating to public health or patient safety.
As outlined in their guidance on RMPs, the EMA requires manufacturers to submit RMPs when applying for a market authorization that are proportionate to the identified risks and the potential risks of the medicinal product, and the need for post-authorization safety data. The EMA also publishes summaries for centrally authorized medicines. The proposed amendments to the FDR would therefore better align Canadian requirements for risk mitigation strategies with those of the EMA by allowing the RMP and the summaries to be submitted in the EMA format.
The EMA allows submissions to be filed for rolling review in emergency situations such as the COVID-19 pandemic.
The EMA requires manufacturers to submit information on the population of study subjects disaggregated by age and sex, and to provide a rationale when this data is not available.
The EMA requires that a drug meet European Pharmacopoeia standards when one exists. They do not require that a list of pharmacopoeia be monitored. Through the proposed amendments, manufacturers that claim a manufacturer’s standard for their drug authorized in Canada would also no longer need to monitor a list of pharmacopoeia, and update their product, if necessary.
The EMA does not require that a manufacturer include the standard of a drug on their label. The proposed amendments would be in alignment with this practice.
United Kingdom
The United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA) has the ability to compel, by law, an authorization holder to conduct tailored activities to manage risks and ensure the benefits of their drugs once on market, similar to the proposed amendments that would permit the Minister to impose T&Cs on all drugs.
MHRA requires sponsors to submit RMPs, RMP summaries and updates for new medicines. They continue to accept the European Union’s versions of an RMP, but may also require an annex including domestic content.
The MHRA released its guidance on rolling review submissions in December 2020 as part of its Brexit policy to streamline the development of novel medicines, including new active substances, as well as biosimilars.
Australia
The Australian Therapeutic Goods Administration (TGA) has the ability to impose conditions at the time a conformity assessment certificate is issued for a medical device, or after the certificate has been issued, similar to the proposed amendments that would permit the Minister to impose T&Cs on medical device licences.
TGA requires RMPs, summaries and updates for specified (higher-risk) products. They continue to accept the European Union’s versions of an RMP, but may also require an annex including domestic content.
The TGA requires that when a standard is available for a drug in one of the default pharmacopoeia outlined in their legislation, a drug at a minimum should meet those standards. Exceptions are considered on a case-by-case basis. With the proposed amendment to the FDR, manufacturers’ standards that differ from those in pharmacopoeia listed under Schedule B would be permitted, if acceptable to the Minister. The limits would need to be qualified with scientific information for review by the Minister as part of a drug submission.
The TGA does not require that a manufacturer include the standard of a drug on their label. The proposed amendments would be in alignment with this practice.
Alignment with other international programs
The amendments to the MDR align Canadian requirements with guidelines outlined by the International Medical Device Regulators Forum (IMDRF) through its continued focus on the device’s safety and effectiveness.
Health Canada is a member of the International Council for Harmonisation. The Council developed a guideline (E2E) describing the fundamental elements of RMPs, which Canada adopted in 2009; Canada thereafter published the associated domestic guidance document in 2015. Outside of opioids, the Canadian system currently relies on guidance alone, while other major jurisdictions (e.g. Europe, Australia) have regulations in place.
Other countries such as Australia, Singapore, Switzerland and the United Kingdom allow submissions to be filed for rolling review during emergency situations.
Health Canada has used rolling reviews to enable collaborative joint reviews of veterinary drugs with the USFDA under the Regulatory Cooperation Council. The proposed regulatory amendments would codify this practice.
With respect to the approach for biologics, Canada is similar to other jurisdictions (the United States, European Union) and the World Health Organization in that it has product-specific rules. Canada’s product-specific rules are overly prescriptive and out-of-date. The proposed regulatory amendments would provide flexibility to keep pace with evolving science and technology over time through outcome-based regulations. Health Canada would continue to rely on international guidelines, like those set out by the Council.
Strategic environmental assessment
In accordance with the Cabinet Directive on the Environmental Assessment of Policy, Plan and Program Proposals, a preliminary scan was conducted which concluded that there will be no expected important environmental effects, either positive or negative; as a result, a detailed analysis is not warranted.
Gender-based analysis plus
Health Canada expects that this proposal would have a positive impact on all individuals in Canada. In some cases, under-representation in clinical trials and unavailability of data in the post-market space could result in specific subpopulations facing disproportionate risks when using particular medical devices and drugs. Depending on the drugs and medical devices that are authorized and the manner in which the proposed regulatory amendments are implemented, there is the potential for some individuals in Canada to receive greater benefit and for some barriers faced by equity-seeking and rights-holding populations (e.g. women and gender diverse individuals, racial and ethnic minorities, persons with disabilities, First Nations, Inuit and Métis) to be addressed.
Terms and conditions for medical devices
The number of people with disability is dramatically increasing due to an aging population and an increase in chronic health conditions.footnote 22,footnote 23 Given that the use of medical devices among individuals experiencing disabilities is disproportionately high as compared to the Canadian population at large,footnote 24 individuals experiencing disability are expected to benefit disproportionately from the proposed amendment to T&Cs for medical devices. Recognizing intersectionality, disability may be more prevalent in certain subpopulations, with a higher prevalence appearing in the elderly, in womenfootnote 25 and particularly in Indigenous women.footnote 26,footnote 27
In addition, race, sex and age could in some cases influence the safety or effectiveness of a medical device. At times, certain population subgroups are underrepresented in the design of a medical device and their associated clinical studies or investigational testing, which could present difficulties in identifying any differences in the safety or effectiveness of a device in those populations. For example, some recent publications have indicated that pulse oximeters may give biased readings of blood oxygen levels on darker skin. Black patients were 29% less likely than white patients to have their need for treatment recognized by the oxygen reader.footnote 28
The proposed amendments to T&Cs for medical devices should lead to increased levels of confidence in the devices used by the diverse populations in Canada.
Disaggregated data, T&Cs and RMPs for drugs
Providing clinical data disaggregated by population subgroups to regulators is a necessary step in identifying potential differences in the effectiveness and safety of a drug within diverse populations. There is growing evidence that the safety and effectiveness of certain health products are impacted by a number of variables, including, among other factors, sex, age and race. For example, certain drugs are metabolized at different rates by male and female populations (e.g. Ambienfootnote 29) and by different racial groups sharing specific genetic traits (e.g. clopidogrel,footnote 30 warfarinfootnote 31. This can result in increased risks for these specific population subgroups, contributing to health inequities in health care.
Furthermore, some subgroup populations remain underrepresented in clinical trials, such as pregnant women, individuals living in rural communities, and the elderly, making it difficult to identify key differences in drug safety or effectiveness within them.
Receiving data that is broken down by population subgroups (e.g. age, sex, race) would allow Health Canada to evaluate the safety and effectiveness of the new drug against these underrepresented populations, where available and appropriate. The proposed amendments would also enable the Department to require the analysis, in the safety specification, of the distribution of risks and uncertainties across relevant subpopulations. If warranted, the Minister could request measures to be included in an RMP. These measures and the safety information received by the Department as a result of these measures could be taken into account by the Minister in their ongoing post-market oversight of the safety of the drug. Measures could include outlining how data would be collected on various patient populations. In addition, T&Cs could be used to require that additional data be collected post-authorization with regard to patient subpopulations for which there is limited clinical data and where the safety or effectiveness of the drug in question needs to be better characterized. The proposed amendments would be important levers to promote increased consideration of diverse subpopulations in clinical trials and for the collection of additional disaggregated data in the post-market space.
Rolling reviews
Two-thirds of those affected by rare diseases are children. Because they affect a relatively small number of patients, there is limited understanding when it comes to their diagnosis and treatment.footnote 32 The proposed option for rolling reviews could contribute to earlier approval of drugs for treatment of such diseases.
In addition, while everyone is susceptible to infectious diseases, certain populations are at risk of more severe outcomes (e.g. older adults, people with chronic medical conditions, and people of any age who are immunocompromised).footnote 33 The proposed rolling review option could contribute to earlier approval of drugs that address an emerging infectious disease and, as a result, would benefit all individuals in Canada, but particularly those subpopulations that are most at risk of severe outcomes and those that are more likely to be exposed to infectious disease.
Standards and modernizing requirements for biologics
It is not anticipated that the changes respecting standards would impact any subpopulations differently. In addition, given that the proposed amendments to modernize the regulations respecting biologic drugs support current practices, they would not be anticipated to impact the general population.
Implementation, compliance and enforcement, and service standards
Implementation
The following proposed regulatory amendments that prioritize burden reduction for industry and ensure that Health Canada is well positioned to address any future public health emergencies would come into force upon registration:
- Amendments related to public health emergency drugs (including T&Cs, rolling reviews and pre-positioning);
- Assuring drug quality during manufacturing;
- Modernizing requirements for biologic drugs;
- Information considered to support the examination of drug submissions;
- Disaggregated clinical trial data for new human drug submissions and supplemental new human drug submissions; and
- Standards.
The following proposed regulatory amendments that require additional time to operationalize would come into force one year after registration:
- T&Cs (for all drugs and for Class II, III or IV medical devices);
- Risk management plans; and
- Rolling reviews (for drugs other than public health emergency drugs).
The following proposed regulatory amendment would come into force at a time set out in a future regulatory amendment once the Department determines that provisions specific to COVID-19 are no longer required:
- Amending the definition of public health emergency drug to no longer include a new drug for which the purpose or conditions of use recommended by the manufacturer relate to COVID-19; and
- Removing other provisions specific to COVID-19.
The Department plans to conduct consultations on the updated key guidance documents that would accompany the proposed regulations at approximately the same time as the Canada Gazette, Part I, prepublication of the regulations. Health Canada would publish updated guidance documents as they are finalized on Canada.ca.
Guidance would provide manufacturers with details of how they may comply with the proposed new provisions.
Compliance and enforcement
Compliance and enforcement of the proposed regulations would be in accordance with a risk-based approach, aligned with existing departmental policies, including compliance promotion and monitoring and enforcement activities in accordance with the Department’s Compliance and enforcement policy for health products (POL-0001).
Health Canada employs a wide range of compliance and enforcement actions and tools. The actions, tools and level of intervention used are dependent on the situation, context and risk to health. Some actions and tools are designed to help regulated parties understand their responsibilities under the law, while other actions and tools are designed to induce compliance with the law. When necessary, enforcement actions are used to address non-compliance with the law. For example, failure to comply with T&Cs would be a contravention to section 21.7 of the Act and could result in the Department taking compliance and enforcement action in accordance with POL-0001 or referring the matter to the Public Prosecution Service of Canada for a potential prosecution.
Contact
Bruno Rodrigue
Executive Director
Office of Legislative and Regulatory Modernization
Policy, Planning and International Affairs Directorate
Health Products and Food Branch
Health Canada
Holland Cross, Suite P2108
11 Holland Avenue
Ottawa, Ontario
K1A 0K9
Address locator: 3000A
Email: lrm.consultations-mlr@hc-sc.gc.ca
PROPOSED REGULATORY TEXT
Notice is given that the Governor in Council proposes to make the annexed Regulations Amending Certain Regulations Made Under the Food and Drugs Act (Agile Licensing) under section 30footnote a of the Food and Drugs Act footnote b.
Interested persons may make representations concerning the proposed Regulations within 100 days after the date of publication of this notice. They are strongly encouraged to use the online commenting feature that is available on the Canada Gazette website but if they use email, mail or any other means, the representations should cite the Canada Gazette, Part I, and the date of publication of this notice, and be sent to Bruno Rodrigue, Executive Director, Office of Legislative and Regulatory Modernization, Health Products and Food Branch, Department of Health, Address Locator: 3000A, 11 Holland Avenue, Suite P2108, Ottawa, Ontario K1A 0K9 (email: lrm.consultations-mlr@hc-sc.gc.ca).
Ottawa, December 6, 2022
Wendy Nixon
Assistant Clerk of the Privy Council
Regulations Amending Certain Regulations Made Under the Food and Drugs Act (Agile Licensing)
Food and Drug Regulations
1 Paragraph A.01.048(d) of the Food and Drug Regulations footnote 34 is replaced by the following:
- (d) sections C.04.001 to C.04.006.
2 (1) Paragraph C.01.004(1)(a) of the Regulations is amended by adding “and” at the end of subparagraph (ii) and by repealing subparagraphs (iii) and (iv).
(2) Subsection C.01.004(5) of the Regulations is replaced by the following:
(5) Subsections (1) to (3) do not apply to
- (a) a drug sold to a drug manufacturer;
- (b) a drug sold under a prescription, if its label carries adequate directions for use and complies with the requirements of section C.01.005; or
- (c) a drug listed in Schedule D to the Act.
3 Section C.01.011 of the Regulations is amended by adding the following after subsection (4):
(5) Subsection (4) does not apply in respect of new drugs, other than those listed in Schedule C to the Act.
4 Section C.01.014.1 of the Regulations is amended by adding the following after subsection (3):
(4) The Minister may request that a manufacturer of a drug for human use, other than a new drug, who has submitted an application under subsection (1) in respect of the drug provide the Minister with a risk management plan for the drug that meets the requirements set out in section C.01.700 if the Minister has reasonable grounds to believe that
- (a) there is a significant degree of uncertainty respecting the risks associated with the drug; or
- (b) the drug presents a serious risk of injury to human health that warrants measures, other than labelling, to reduce the probability or severity of such an injury.
5 Subsection C.01.014.2(2) of the Regulations is replaced by the following:
(2) The Minister may refuse to issue the document if
- (a) the Minister has reasonable grounds to believe that the product to which the application for the drug identification number relates is not a drug;
- (b) the Minister has reasonable grounds to believe that the sale of the drug would
- (i) cause injury to the health of the purchaser or consumer, or
- (ii) contravene the Act or these Regulations; or
- (c) in the case where the Minister has requested a risk management plan for the drug under subsection C.01.014.1(4), the manufacturer fails to provide the Minister with a risk management plan that meets the requirements set out in section C.01.700.
6 (1) Subsection C.01.014.21(1.1) of the Regulations is replaced by the following:
(1.1) The Minister may, at any time, impose terms and conditions on a drug identification number assigned for a public health emergency drug, or amend those terms and conditions, if
- (a) a notice of compliance was issued under section C.08.004 in respect of
- (i) a new drug submission that was filed under section C.08.002 for the public health emergency drug that contains the statement referred to in paragraph C.08.002(2.1)(a), or
- (ii) a supplement to a new drug submission referred to in subparagraph (i) that was filed under section C.08.003 for the public health emergency drug; or
- (b) a notice of compliance was issued under section C.08.004 in respect of one of the following submissions or supplements that was filed for the drug on the basis of a direct or indirect comparison to another public health emergency drug referred to in paragraph (a):
- (i) a new drug submission under section C.08.002,
- (ii) an abbreviated new drug submission under section C.08.002.1, or
- (iii) a supplement to a new drug submission or abbreviated new drug submission under section C.08.003.
(2) The definition designated COVID-19 drug in subsection C.01.014.21(3) of the Regulations is repealed.
(3) Subsection C.01.014.21(3) of the Regulations is amended by adding the following in alphabetical order:
- public health emergency drug
- has the same meaning as in section C.08.001.1. (drogue d’urgence de santé publique)
(4) Section C.01.014.21 of the Regulations is replaced by the following:
C.01.014.21 The Minister may, at any time, impose terms and conditions on a drug identification number assigned for a drug, or amend those terms and conditions, after considering the following factors:
- (a) whether there are significant uncertainties relating to the benefits or risks associated with the drug;
- (b) whether the requirements under the Act are sufficient to
- (i) optimize the benefits and manage the risks associated with the drug,
- (ii) manage the uncertainties relating to the benefits and risks, and
- (iii) collect information to be able to continuously assess the benefits and risks, identify any changes to them and manage the uncertainties;
- (c) whether the proposed terms and conditions may contribute to meeting the objectives set out in subparagraphs (b)(i) to (iii);
- (d) whether compliance with the proposed terms and conditions is technically feasible; and
- (e) whether there are less burdensome ways to meet the objectives of the proposed terms and conditions.
7 The Regulations are amended by adding the following after section C.01.625:
Risk Management Plans
C.01.700 A risk management plan that is provided to the Minister under these Regulations must take into account the Canadian context and include
- (a) a description of the drug to which the plan relates and the uses of the drug;
- (b) a detailed description of the risks associated with the drug and the uncertainties relating to those risks;
- (c) a detailed description of the measures that the manufacturer intends to take to address and monitor the uncertainties;
- (d) a detailed description of the measures that the manufacturer intends to take to prevent or reduce the risks;
- (e) a detailed description of how the manufacturer intends to evaluate the effectiveness of the measures that they intend to take to prevent or reduce the risks; and
- (f) a summary of the plan’s contents, in English and in French.
C.01.701 (1) The Minister may request that the manufacturer of a drug for human use for which a drug identification number has been assigned and has not been cancelled provide the Minister with a risk management plan for the drug that meets the requirements set out in section C.01.700 if
- (a) a risk management plan for the drug has not yet been provided to the Minister; and
- (b) the Minister has reasonable grounds to believe that
- (i) there is a significant degree of uncertainty respecting the risks associated with the drug, or
- (ii) the drug presents a serious risk of injury to human health that warrants measures, other than labelling, to reduce the probability or severity of such an injury.
(2) If the manufacturer fails, within the time specified in the request, to provide the Minister with a risk management plan for the drug that meets the requirements set out in section C.01.700, the manufacturer shall not sell the drug until they provide the Minister with such a plan.
C.01.702 A manufacturer of a drug for human use for which a drug identification number has been assigned and has not been cancelled shall, as soon as feasible, provide the Minister with an updated risk management plan for the drug that meets the requirements set out in section C.01.700 if
- (a) the risks associated with the drug, or the uncertainties relating to those risks, are significantly different than those that are described in the existing plan; or
- (b) the measures that the manufacturer intends to take to address and monitor the uncertainties relating to the risks associated with the drug or to prevent or reduce those risks are significantly different than those that are described in the existing plan.
C.01.703 (1) The Minister may request that the manufacturer of a drug for human use for which a drug identification number has been assigned and has not been cancelled provide the Minister with an updated risk management plan for the drug that meets the requirements set out in section C.01.700 if, on the basis of information obtained after the existing plan was provided to the Minister, the Minister has reasonable grounds to believe that
- (a) the risks associated with the drug, or the uncertainties relating to those risks, are significantly different than those that are described in the existing plan; or
- (b) the drug presents a serious risk of injury to human health that warrants measures to reduce the probability or severity of such an injury that are significantly different than those that are described in the existing plan.
(2) If the manufacturer fails, within the time specified in the request, to provide the Minister with an updated risk management plan for the drug that meets the requirements set out in section C.01.700, the manufacturer shall not sell the drug until they provide the Minister with such a plan.
8 (1) Subsection C.01A.005(2) of the Regulations is replaced by the following:
(2) An application for an establishment licence that relates to one or more activities set out in Table I to section C.01A.008 to be carried out in respect of a category of drugs set out in Table II to that section that includes one or more of the following drugs may include a statement to that effect:
- (a) a COVID-19 drug; or
- (b) a drug that is manufactured, sold or represented for use in relation to a condition described in the List of Conditions that Threaten Public Health, as defined in section C.08.001.1.
(2) Subsection C.01A.005(2) of the Regulations is replaced by the following:
(2) An application for an establishment licence that relates to one or more activities set out in Table I to section C.01A.008 to be carried out in respect of a category of drugs set out in Table II to that section that includes a drug that is manufactured, sold or represented for use in relation to a condition described in the List of Conditions that Threaten Public Health, as defined in section C.08.001.1, may include a statement to that effect.
9 (1) Subsection C.01A.006(1.1) of the Regulations is replaced by the following:
(1.1) An application to amend an establishment licence that relates to one or more activities set out in Table I to section C.01A.008 to be carried out in respect of a category of drugs set out in Table II to that section that includes one or more of the following drugs may include a statement to that effect:
- (a) a COVID-19 drug; or
- (b) a drug that is manufactured, sold or represented for use in relation to a condition described in the List of Conditions that Threaten Public Health, as defined in section C.08.001.1.
(2) Subsection C.01A.006(1.1) of the Regulations is replaced by the following:
(1.1) An application to amend an establishment licence that relates to one or more activities set out in Table I to section C.01A.008 to be carried out in respect of a category of drugs set out in Table II to that section that includes a drug that is manufactured, sold or represented for use in relation to a condition described in the List of Conditions that Threaten Public Health, as defined in section C.08.001.1, may include a statement to that effect.
10 (1) Subsection C.01A.008(1.1) of the Regulations is replaced by the following:
(1.1) The Minister shall, in determining whether he or she has received the information and material referred to in sections C.01A.005 to C.01A.007 in relation to an application referred to in subsection C.01A.005(2) or C.01A.006(1.1) that contains the statement referred to in the applicable subsection, also take into consideration the public health need related to COVID-19 or the condition described in the List of Conditions that Threaten Public Health, as defined in section C.08.001.1, as the case may be.
(2) Subsection C.01A.008(1.1) of the Regulations is replaced by the following:
(1.1) The Minister shall, in determining whether he or she has received the information and material referred to in sections C.01A.005 to C.01A.007 in relation to an application referred to in subsection C.01A.005(2) or C.01A.006(1.1) that contains the statement referred to in the applicable subsection, also take into consideration the public health need related to the condition described in the List of Conditions that Threaten Public Health, as defined in section C.08.001.1.
(3) Section C.01A.008 of the Regulations is amended by adding the following after subsection (4):
(5) If an application for or to amend an establishment licence contains, in accordance with subsection C.01A.005(2) or C.01A.006(1.1), a statement that refers to a condition that is subsequently removed from the List of Conditions that Threaten Public Health, as defined in section C.08.001.1, the Minister may nonetheless issue or amend the licence on the basis of the application.
11 Section C.01A.012.1 of the Regulations is amended by adding the following after subsection (2):
(3) For greater certainty, the power in subsection (1) continues to apply in respect of an establishment licence until the licence is cancelled, even if the holder does not conduct activities in respect of the drug that formed the basis for the statement contained in the application that led to the issuance or amendment of the licence.
12 The heading before section C.02.013 of the Regulations is replaced by the following:
Quality Control
C.02.012.1 Every lot or batch of a drug shall be fabricated, packaged/labelled, tested and stored, including during transportation, in a manner that assures the quality of the drug.
13 Subsection C.02.019(4.1) of the Regulations is replaced by the following:
(4.1) Subsections (1) and (2) do not apply to a distributor or importer of a COVID-19 drug if the lot of the drug is the subject of a request made under subsection C.04.007(1).
14 Section C.03.206 of the Regulations is replaced by the following:
C.03.206 Sections C.01.005 and C.04.009 do not apply to a component or kit.
15 Division 4 of Part C of the Regulations is replaced by the following:
DIVISION 4
Schedule D Drugs
Definitions
C.04.001 The following definitions apply in this Division.
- biological source material
- means
- (a) biological material sourced or derived from humans;
- (b) animals, including insects, or any biological material sourced or derived from them;
- (c) plants or any biological material sourced or derived from them; or
- (d) micro-organisms, including bacteria, viruses, fungi and bacteriophages, or any biological material sourced or derived from them. (matériel d’origine biologique)
- drug
- means a drug that is listed in Schedule D to the Act that is in dosage form or an active ingredient that can be used in the preparation of a drug listed in that Schedule. (drogue)
- holder,
- in respect of a drug identification number, means the manufacturer to whom the document setting out the number was issued under subsection C.01.014.2(1). (titulaire)
Prohibitions on Sale
C.04.002 It is prohibited for a distributor or importer of a drug to sell the drug unless the drug has been fabricated, packaged/labelled and tested in accordance with this Division.
C.04.003 It is prohibited for a person to sell a drug that they have fabricated, packaged/labelled or tested unless they have fabricated, packaged/labelled or tested it, as the case may be, in accordance with this Division.
Biological Source Material
C.04.004 (1) It is prohibited for a person to use biological source material in the fabrication of a drug unless
- (a) the material is
- (i) prepared and stored in a manner that ensures its suitability for use in the fabrication of the drug, and
- (ii) collected, in the case of material of human or animal origin, under medical or veterinary supervision, as the case may be;
- (b) the person collects the information that is necessary to allow the tracing of the material; and
- (c) any human from whom, or animal from which, the material is collected — or any animal that is such material — is free from any disease that would make the material unsuitable for use.
(2) A person who uses biological source material in the fabrication of a drug must determine a retention period for the information referred to in paragraph (1)(b), taking into account
- (a) the nature of the material;
- (b) the risks associated with the material;
- (c) how the material is used in fabrication of the drug; and
- (d) in the case where the material is used in the fabrication of a drug in dosage form, how the drug is used.
(3) A person who uses biological source material in the fabrication of a drug must retain the information referred to in paragraph (1)(b) for at least the longer of
- (a) five years after the biological source material was last used in the fabrication of the drug, and
- (b) the period determined under subsection (2).
(4) A person who uses biological source material in the fabrication of a drug must ensure that the material meets any specifications for the material that have been provided to the Minister in connection with the drug.
Prevention of Contamination
C.04.005 (1) Every person who fabricates a drug and every person who packages a drug in an immediate container must
- (a) segregate all work with infectious agents that require special handling, including spore-bearing pathogenic micro-organisms; and
- (b) minimize the possibility of contamination of biological source material and drugs at the premises where the fabrication or packaging of the drug takes place, including by taking measures to protect any individual who has access to the area where the fabrication or packaging takes place against infection.
(2) It is prohibited for a person to conduct laboratory procedures of a diagnostic nature in their premises unless those procedures are segregated from the fabrication, packaging/labelling and testing of drugs.
Reference Preparations
C.04.006 Reference preparations that are used to test the purity or potency of a drug must be adequate to control the quality of the drug.
Lot Release
C.04.007 (1) The Minister may, for the purpose of assessing whether a lot of a drug in dosage form is suitable for sale, request that a fabricator, packager/labeller or importer of the drug, or the holder of the drug identification number, provide the Minister with information, samples of the drug or of its active ingredients or material to be used to test the samples.
(2) It is prohibited for a person who is requested to provide information, samples or material under subsection (1) — and any person whom the Minister notifies of the request — to sell drugs from the lot to which the request relates unless the Minister notifies the person that the lot is suitable for sale.
(3) In this section, suitable for sale means that the lot has been fabricated, packaged/labelled and tested in accordance with these Regulations and in a manner that is consistent with information that has been provided to the Minister regarding the quality and safety of the drug.
Periodic Quality Reporting
C.04.008 The holder of the drug identification number for a drug in dosage form must, at the request of the Minister, provide the Minister, on an annual basis or any longer interval specified by the Minister, with information regarding the quality of the drug and its active ingredients, including information regarding the consistency of the fabrication and packaging processes for the drug and the ingredients.
Labelling
C.04.009 (1) The principal display panel of both the inner and outer labels of a drug in dosage form must show
- (a) the drug’s proper name, if any, which, if there is a brand name, must immediately precede or follow the brand name in type not less than one-half the size of that of the brand name;
- (b) if there is no proper name, the drug’s common name;
- (c) the net quantity of the drug in the package;
- (d) if these Regulations require the drug to be sterile, the notations “sterile” and “stérile”;
- (e) if the biological source material used in the fabrication of the drug or its active ingredients was sourced or derived from humans, an indication to that effect; and
- (f) if the biological source material used in the fabrication of the drug or its active ingredients was sourced or derived from animals, the species of origin.
(2) The inner and outer labels of a drug in dosage form must show on any panel
- (a) the name of the holder of the drug identification number for the drug;
- (b) the potency of the drug, if applicable;
- (c) the recommended dose of the drug;
- (d) the lot number of the drug;
- (e) subject to subsection (7), the expiration date of the drug;
- (f) subject to subsection (3), directions for use; and
- (g) subject to subsection (4), any other information that is necessary to prevent injury to human health.
(3) Paragraph (2)(f) does not apply if directions for use are required to be displayed on the label under section C.01.004.02 or C.01.004.03.
(4) Despite paragraph (2)(g), if another provision of these Regulations requires that information referred to in that paragraph be shown on a particular panel of a label, the information must be shown on that panel.
(5) The outer label of a drug in dosage form must show on any panel
- (a) the address of the holder of the drug identification number for the drug;
- (b) a quantitative list of any preservatives contained in the drug, by their proper names, or if a preservative has no proper name, by its common name;
- (c) the approved storage conditions for the drug;
- (d) any templates or other information that is necessary to support proper storage and handling of the drug; and
- (e) in the case of a new drug for extraordinary use in respect of which a notice of compliance has been issued under section C.08.004.01, the following statement, displayed in capital letters and in a legible manner:
“HEALTH CANADA HAS AUTHORIZED THE SALE OF THIS EXTRAORDINARY USE NEW DRUG FOR [naming purpose] BASED ON LIMITED CLINICAL TESTING IN HUMANS.
SANTÉ CANADA A AUTORISÉ LA VENTE DE CETTE DROGUE NOUVELLE POUR USAGE EXCEPTIONNEL AUX FINS DE [indication de la fin] EN SE FONDANT SUR DES ESSAIS CLINIQUES RESTREINTS CHEZ L’ÊTRE HUMAIN.”.
(6) If the immediate container of a drug in dosage form is too small to accommodate an inner label that meets the requirements of these Regulations, the inner label requirements of these Regulations do not apply if
- (a) there is an outer label that meets the requirements of these Regulations; and
- (b) the inner label shows
- (i) the drug’s proper name or, if there is no proper name, its common name or, in the case of a drug that contains more than one active ingredient, its brand name,
- (ii) the name of the holder of the drug identification number for the drug,
- (iii) the potency of the drug, if applicable, unless the drug contains more than one active ingredient and the brand name of the drug is unique for a particular potency of the drug,
- (iv) the lot number of the drug,
- (v) subject to subsection (7), the expiration date of the drug,
- (vi) the route of administration of the drug,
- (vii) any information that is necessary to prevent injury to human health, and
- (viii) the net quantity of the drug in the container.
(7) The expiration date referred to in paragraph (2)(e) and subparagraph (6)(b)(v) may be omitted from the label of a drug that is stockpiled for use in emergency situations if an alternative means is provided to communicate the date to the individuals who administer the drug.
Labelling — Prescription Drugs
C.04.010 (1) Every package of a drug in dosage form that is a prescription drug must carry the symbol “Pr” on the upper left quarter of the principal display panel of both the inner and outer labels or, in the case of a single-dose container, on the upper left quarter of the principal display panel of the outer label.
(2) Subsection (1) does not apply to
- (a) drugs sold to a person who holds an establishment licence under Division 1A; and
- (b) drugs sold under a prescription.
16 (1) The definition designated COVID-19 drug in section C.08.001.1 of the Regulations is repealed.
(2) Section C.08.001.1 of the Regulations is amended by adding the following in alphabetical order:
- List of Conditions that Threaten Public Health
- means the List of Conditions that Threaten Public Health in Canada, published on the Government of Canada’s website, as amended from time to time; (Liste d’affections qui menacent la santé publique)
- public health emergency drug
- means a new drug for which the purpose and conditions of use recommended by the manufacturer relate to COVID-19 or a condition that is described in the List of Conditions that Threaten Public Health; (drogue d’urgence de santé publique)
(3) The definition public health emergency drug in section C.08.001.1 of the Regulations is replaced by the following:
- public health emergency drug
- means a new drug for which the purpose and conditions of use recommended by the manufacturer relate to a condition that is described in the List of Conditions that Threaten Public Health; (drogue d’urgence de santé publique)
17 The Regulations are amended by adding the following after section C.08.001.1:
C.08.001.2 The Minister may add a condition to the List of Conditions that Threaten Public Health only if the Minister has reasonable grounds to believe that
- (a) the condition presents, or is the result of, a significant risk to public health in Canada; and
- (b) immediate action is required to deal with the risk.
C.08.001.3 If a condition is removed from the List of Conditions that Threaten Public Health after a new drug submission for a public health emergency drug that relates to the condition is filed in accordance with subsections C.08.002(2) to (2.5) and section C.08.005.1 but before the Minister makes a final decision in respect of the submission under section C.08.004, the drug remains a public health emergency drug for the purposes of this Division until such a decision is made.
18 (1) Paragraph C.08.002(2)(o) of the Regulations is replaced by the following:
- (o) in the case of a new drug for human use other than a public health emergency drug, an assessment as to whether there is a likelihood that the new drug will be mistaken for another drug for which a drug identification number has been assigned due to a resemblance between the brand name that is proposed to be used in respect of the new drug and the brand name, common name or proper name of the other drug.
(2) Subsection C.08.002(2) of the Regulations is amended by striking out “and” at the end of paragraph (n), by adding “and” at the end of paragraph (o) and by adding the following after paragraph (o):
- (p) in the case of a new drug for human use, a risk management plan for the drug that meets the requirements set out in section C.01.700 if
- (i) there is a significant degree of uncertainty respecting the risks associated with the drug, or
- (ii) the drug presents a serious risk of injury to human health that warrants measures, other than labelling, to reduce the probability or severity of such an injury.
(3) Subsections C.08.002(2.1) to (2.5) of the Regulations are replaced by the following:
(2.01) If clinical trial data that is included in a new drug submission under paragraph (2)(g) or (h) or (2.1)(b) is broken down by population subgroup in an application made to the European Medicines Agency or the United States Food and Drug Administration to authorize the sale of the drug, the data must be broken down in the same manner in the new drug submission.
(2.1) A manufacturer may file, for a public health emergency drug, a new drug submission that does not meet the requirements set out in paragraphs (2)(g) and (h) if the submission contains
- (a) a statement that the submission contains the evidence referred to in paragraph (b); and
- (b) sufficient evidence to support the conclusion that the benefits associated with the drug outweigh the risks associated with it for the purpose and under the conditions of use recommended, with consideration given to the uncertainties relating to those benefits and risks as well as the public health need related to COVID-19 or the applicable condition described in the List of Conditions that Threaten Public Health, as the case may be.
(2.2) A manufacturer may file, for a public health emergency drug for human use, a new drug submission that does not meet the requirements set out in paragraph (2)(j.1) if the submission contains a draft of every label to be used in connection with the drug, including any package insert and any document that is provided on request and that sets out supplementary information on the use of the drug.
(2.3) If, at the time a new drug submission is filed for a public health emergency drug, the manufacturer is unable to provide the Minister with information or material that is required under any of paragraphs (2)(e) to (k), (m) and (n), paragraph (2.1)(b), subsection (2.2) or section C.08.005.1 or any of that information or material is incomplete, the manufacturer shall provide the Minister, at that time, with a plan that specifies how and when they will provide the Minister with the missing information or material.
(2.4) Subsections (2.1) to (2.3) apply only if
- (a) the new drug submission contains a statement that the submission is for a public health emergency drug; and
- (b) the purpose and conditions of use specified in the new drug submission relate only to COVID-19 or a condition described in the List of Conditions that Threaten Public Health and the submission contains a statement to that effect.
(2.5) Subsections (2.1) to (2.3) do not apply if the manufacturer is seeking a notice of compliance for a public health emergency drug on the basis of a direct or indirect comparison between the public health emergency drug and another public health emergency drug.
(4) Paragraph C.08.002(2.1)(b) of the Regulations is replaced by the following:
- (b) sufficient evidence to support the conclusion that the benefits associated with the drug outweigh the risks associated with it for the purpose and under the conditions of use recommended, with consideration given to the uncertainties relating to those benefits and risks as well as the public health need related to the applicable condition described in the List of Conditions that Threaten Public Health.
(5) Subsection C.08.002(2.3) of the Regulations is replaced by the following:
(2.3) If, at the time a new drug submission is filed for a public health emergency drug, the manufacturer is unable to provide the Minister with information or material that is required under any of paragraphs (2)(e) to (k), (m), (n) and (p), paragraph (2.1)(b), subsection (2.2) or section C.08.005.1 or any of that information or material is incomplete, the manufacturer shall provide the Minister, at that time, with a plan that specifies how and when they will provide the Minister with the missing information or material.
(6) Paragraph C.08.002(2.4)(b) of the Regulations is replaced by the following:
- (b) the purpose and conditions of use specified in the new drug submission relate only to a condition described in the List of Conditions that Threaten Public Health and the submission contains a statement to that effect.
(7) Section C.08.002 of the Regulations is amended by adding the following after subsection (3):
(4) A manufacturer may file a new drug submission that does not yet contain all of the information that is required under paragraphs (2)(g) to (i), (m), (n) and (p) — and, if applicable, all of the corresponding information that is required under section C.08.005.1 — if
- (a) prior to filing the submission, the manufacturer provides the Minister with
- (i) information that demonstrates that the conditions set out in any of paragraphs (5)(a) to (c) are met,
- (ii) a plan that specifies how and when, within the 155-day period that begins on the day on which the submission is filed, the manufacturer proposes to provide the Minister with the missing information,
- (iii) information that demonstrates that the manufacturer possesses a significant amount of evidence to establish the safety of the new drug for the purpose and under the conditions of use recommended,
- (iv) information that demonstrates that the manufacturer possesses a significant amount of evidence of the clinical effectiveness of the new drug for the purpose and under the conditions of use recommended, and
- (v) in the case where the notice of compliance will be sought on the basis of a direct or indirect comparison to a drug for which a drug identification number has been assigned, the drug identification number;
- (b) the Minister provides the manufacturer with a notice that specifies how and when, within the 155-day period referred to in subparagraph (a)(ii), the missing information is to be provided; and
- (c) the submission
- (i) contains the evidence referred to in subparagraphs (a)(iii) and (iv), and
- (ii) is filed within 60 days after the day on which the Minister provides the manufacturer with the notice.
(5) For the purposes of subparagraphs (4)(a)(i) and C.08.003(5)(a)(i), the conditions are the following:
- (a) the new drug is needed to diagnose, treat, mitigate or prevent an emerging infectious disease that poses or may pose a serious risk of injury to human or animal health;
- (b) the new drug is intended to diagnose, treat, mitigate or prevent a disease, disorder or abnormal physical state that poses or may pose a serious risk of injury to human or animal health and the recommended purpose and conditions of use of the drug do not fall within the recommended purposes and conditions of use of any other drug for which a drug identification number has been assigned and has not been cancelled; and
- (c) the new drug is intended to diagnose, treat, mitigate or prevent a disease, disorder or abnormal physical state that poses or may pose a serious risk of injury to human or animal health and the recommended purpose and conditions of use of the drug fall within the recommended purposes and conditions of use of one or more other drugs for which a drug identification number has been assigned and has not been cancelled, but there are reasonable grounds to believe that the new drug is significantly more effective, or poses a significantly lower risk, than each of the other drugs.
(6) The Minister shall provide the manufacturer with the notice referred to in paragraph (4)(b) if
- (a) the manufacturer has provided the Minister with the necessary information in accordance with paragraph (4)(a); and
- (b) the Minister has no reason to believe that the manufacturer will be unable to provide the missing information in accordance with the notice.
(7) A new drug submission that is filed in accordance with subsection (4) is considered to have been cancelled by the manufacturer if
- (a) the manufacturer has failed, or will be unable, to provide information specified in the notice referred to in paragraph (4)(b) to the Minister within 10 days after the relevant date specified in the notice or any longer period specified by the Minister; or
- (b) the notice of compliance is being sought on the basis of a direct or indirect comparison to a drug for which a drug identification number has been assigned and the manufacturer failed to provide the number to the Minister before filing the submission.
(8) A manufacturer of a new drug for veterinary use only may file a new drug submission for that drug that does not yet contain all of the information that is required under paragraphs (2)(d) to (j), subparagraphs (2)(k)(ii) and (iv) and paragraphs (2)(m) and (n) — and, if applicable, all of the corresponding information that is required under section C.08.005.1 — if
- (a) the Minister has indicated to the manufacturer an intent to conduct a review of the submission with a foreign regulatory authority, as defined in subsection C.10.001(1); and
- (b) at the time of filing, the submission contains
- (i) a plan that specifies how and when the manufacturer will provide the Minister with the missing information,
- (ii) at least some of the information that is required under any of paragraphs (2)(d) to (h), (m) and (n), and
- (iii) in the case where the notice of compliance is being sought on the basis of a direct or indirect comparison to a drug for which a drug identification number has been assigned, the drug identification number.
(9) A new drug submission that is filed in accordance with subsection (8) is considered to have been cancelled by the manufacturer if
- (a) the manufacturer notifies the Minister that they no longer wish to have the Minister conduct the review of the submission with the foreign regulatory authority; or
- (b) the notice of compliance is being sought on the basis of a direct or indirect comparison to a drug for which a drug identification number has been assigned and the manufacturer failed to provide the number to the Minister when the submission was filed.
19 Paragraph C.08.002.01(2)(b) of the Regulations is amended by striking out “and” at the end of subparagraph (ix), by adding “and” at the end of subparagraph (x) and by adding the following after subparagraph (x):
- (xi) a risk management plan for the new drug that meets the requirements set out in section C.01.700.
20 Subsection C.08.002.1(2) of the Regulations is amended by adding the following after paragraph (a):
- (a.1) in the case of a new drug for human use, a risk management plan for the drug that meets the requirements set out in section C.01.700 if either of the conditions set out in subparagraphs C.08.002(2)(p)(i) and (ii) is met;
21 (1) Subsection C.08.003(2) of the Regulations is amended by adding the following after paragraph (b):
- (b.1) in the case of an influenza vaccine that is referred to in the List of Influenza Vaccines for Which Supplements to New Drug Submissions Can Be Filed, published on the Government of Canada’s website, as amended from time to time, the strains to which the vaccine relates;
(2) Section C.08.003 of the Regulations is amended by adding the following after subsection (3):
(3.01) If clinical trial data that is included in a supplement to a new drug submission is broken down by population subgroup in an application made to the European Medicines Agency or the United States Food and Drug Administration to authorize the sale of the drug, the data must be broken down in the same manner in the supplement.
(3) Section C.08.003 of the Regulations is amended by adding the following after subsection (4):
(5) A manufacturer of a new drug for which a new drug submission was filed may file a supplement to that submission that does not yet contain all of the information and material that is required under subsection (3) — and, if applicable, all of the corresponding information that is required under section C.08.005.1 — if
- (a) prior to filing the supplement, the manufacturer provides the Minister with
- (i) information that demonstrates that the conditions set out in any of paragraphs C.08.002(5)(a) to (c) are met,
- (ii) a plan that specifies how and when, within the 155-day period that begins on the day on which the supplement is filed, they propose to provide the Minister with the missing information or material,
- (iii) information that demonstrates that the manufacturer possesses a significant amount of the information and material that is required under subsection (3), and
- (iv) in the case where the notice of compliance will be sought on the basis of a direct or indirect comparison to a drug for which a drug identification number has been assigned, the drug identification number;
- (b) the Minister provides the manufacturer with a notice that specifies how and when, within the 155-day period referred to in subparagraph (a)(ii), the missing information or material is to be provided; and
- (c) the supplement
- (i) contains the information and material that is required under subsection (3) that was in the manufacturer’s possession when they provided information under subparagraph (a)(iii), and
- (ii) is filed within 60 days after the day on which the Minister provides the manufacturer with the notice.
(6) The Minister shall provide the manufacturer with the notice referred to in paragraph (5)(b) if
- (a) the manufacturer has provided the Minister with the necessary information in accordance with paragraph (5)(a); and
- (b) the Minister has no reason to believe that the manufacturer will be unable to provide the missing information or material in accordance with the notice.
(7) A supplement that is filed in accordance with subsection (5) is considered to have been cancelled by the manufacturer if
- (a) the manufacturer has failed, or will be unable, to provide information or material specified in the notice referred to in paragraph (5)(b) to the Minister within 10 days after the relevant date specified in the notice or any longer period specified by the Minister; or
- (b) the notice of compliance is being sought on the basis of a direct or indirect comparison to a drug for which a drug identification number has been assigned and the manufacturer failed to provide the number to the Minister before filing the supplement.
(8) A manufacturer of a new drug for veterinary use only for which a new drug submission was filed may file a supplement to that submission that does not yet contain all of the information and material that is required under subsection (3) — and, if applicable, all of the corresponding information that is required under section C.08.005.1 — if
- (a) the Minister has indicated to the manufacturer an intent to conduct a review of the supplement with a foreign regulatory authority, as defined in subsection C.10.001(1); and
- (b) at the time of filing, the supplement contains
- (i) a plan that specifies how and when the manufacturer will provide the Minister with the missing information or material,
- (ii) some of the information or material that is required under subsection (3), and
- (iii) in the case where the notice of compliance is being sought on the basis of a direct or indirect comparison to a drug for which a drug identification number has been assigned, the drug identification number.
(9) A supplement that is filed in accordance with subsection (8) is considered to have been cancelled by the manufacturer if
- (a) the manufacturer notifies the Minister that they no longer wish to have the Minister conduct the review of the supplement with the foreign regulatory authority; or
- (b) the notice of compliance is being sought on the basis of a direct or indirect comparison to a drug for which a drug identification number has been assigned and the manufacturer failed to provide the number to the Minister when the supplement was filed.
(10) A manufacturer of a vaccine referred to in the list referred to in paragraph (2)(b.1) may file a supplement to the new drug submission for the vaccine that does not yet contain all of the information and material that is required under subsection (3) — and, if applicable, all of the corresponding information that is required under section C.08.005.1 — if the supplement relates to the matter specified in that paragraph.
22 Section C.08.003.1 of the Regulations is replaced by the following:
C.08.003.1 In examining a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission, an abbreviated extraordinary use new drug submission or a supplement to any of those submissions, the Minister may, for the purpose of assessing the safety and effectiveness of the new drug for which the submission or supplement has been filed, examine
- (a) information or material provided by any person under the Act;
- (b) information or material obtained from sites at which the new drug or any active ingredient of the drug, as defined in subsection C.01A.001(1), is or is proposed to be manufactured, packaged, labelled or tested; and
- (c) information or material obtained, directly or indirectly, from a foreign regulatory authority, as defined in subsection C.10.001(1).
23 The Regulations are amended by adding the following after section C.08.003.1:
C.08.003.2 For greater certainty, the obligation to provide a risk management plan under paragraph C.08.002(2)(p), subparagraph C.08.002.01(2)(b)(ix) or paragraph C.08.002.1(2)(a.1) may arise at any time before the Minister makes a final decision under section C.08.004 or C.08.004.01, as the case may be.
24 The heading before section C.08.009.01 of the Regulations is replaced by the following:
Pre-positioning of Public Health Emergency Drugs
25 Section C.08.009.01 of the Regulations is amended by adding the following in alphabetical order:
- dosage form class
- has the same meaning as in subsection C.01A.001(1). (classe de forme posologique)
- fabricate
- has the same meaning as in subsection C.01A.001(1). (manufacturer)
- package/label
- has the same meaning as in subsection C.01A.001(1). (emballer-étiqueter)
26 Section C.08.009.02 of the Regulations is replaced by the following:
C.08.009.02 Sections C.08.009.03 to C.08.009.05 apply in respect of a public health emergency drug if
- (a) a notice of compliance has not been issued in respect of the drug under section C.08.004 or C.08.004.01; and
- (b) His Majesty in right of Canada has entered into a contract for the procurement of the drug.
27 Subsection C.08.009.03(1) of the Regulations is replaced by the following:
C.08.009.03 (1) The holder of an establishment licence may import a public health emergency drug if the following conditions are met:
- (a) the Chief Public Health Officer provides the Minister with
- (i) information indicating that the drug is the subject of a new drug submission or an application made to a foreign regulatory authority to authorize its sale,
- (ii) the name of the drug and a description of it,
- (iii) the name and contact information of the drug’s manufacturer,
- (iv) information specifying the quantity of the drug to be imported,
- (v) the name and contact information of the holder, and
- (vi) the civic address of the place where the drug will be stored after it is imported;
- (b) the holder provides the Minister with
- (i) the name and contact information of each fabricator, packager/labeller and tester of the drug and the civic address of each building at which the drug will be fabricated, packaged/labelled or tested, specifying for each building
- (A) the activities referred to in Table I to section C.01A.008 that apply to the drug,
- (B) the categories referred to in Table II to that section that apply to the drug, and
- (C) for each category, the dosage form classes, if any, and whether the drug will be in a sterile form, and
- (ii) a certificate from an inspector indicating that each fabricator’s, packager/labeller’s and tester’s buildings, equipment, practices and procedures meet the applicable requirements of Divisions 2 to 4 or, alternatively, other evidence establishing that those requirements are met; and
- (i) the name and contact information of each fabricator, packager/labeller and tester of the drug and the civic address of each building at which the drug will be fabricated, packaged/labelled or tested, specifying for each building
- (c) the drug is part of a category of drugs that are authorized to be imported under the licence.
28 (1) The portion of section C.08.009.04 of the Regulations before paragraph (a) is replaced by the following:
C.08.009.04 Sections A.01.040, C.01.004.1 and C.01A.006 and Divisions 2 to 4, other than the following provisions, do not apply in respect of the importation of a public health emergency drug under section C.08.009.03:
(2) Section C.08.009.04 of the Regulations is amended by adding the following after paragraph (b):
- (b.1) section C.02.012.1, as it applies to the storage of the drug by the holder;
(3) Section C.08.009.04 of the Regulations is amended by striking out “and” after paragraph (i), by adding “and” after paragraph (h) and by repealing paragraph (j).
29 The portion of section C.08.009.05 of the Regulations before paragraph (a) is replaced by the following:
C.08.009.05 Despite anything in these Regulations, the holder of an establishment licence may distribute a public health emergency drug that they have imported under section C.08.009.03 if
30 (1) Subsection C.08.011.2(2) of the Regulations is amended by adding the following after paragraph (c):
- (c.1) section C.02.012.1, as it applies to the storage of the new drug by the holder of an establishment licence;
(2) Subsection C.08.011.2(2) of the Regulations is amended by striking out “and” after paragraph (j), by adding “and” after paragraph (i) and by repealing paragraph (k).
31 (1) Subsection C.10.001(5) of the Regulations is amended by adding the following after paragraph (c):
- (c.1) section C.02.012.1 as it applies to the storage of the drug by the licensee;
(2) Subsection C.10.001(5) of the Regulations is by striking out “and” after paragraph (j), by adding “and” after paragraph (i) and by repealing paragraph (k).
32 Subsection C.10.002(2) of the Regulations is amended by adding the following after paragraph (c):
- (c.1) section C.02.012.1 as it applies to the storage of the drug by the licensee;
33 The Regulations are amended by replacing “designated COVID-19 drug” with “drug” in the following provisions:
- (a) paragraphs C.08.009.04(a), (d) and (e); and
- (b) paragraphs C.08.009.05(a) and (b).
Medical Devices Regulations
34 Subsections 36(2) to (4) of the Medical Devices Regulations footnote 35 are replaced by the following:
(2) The Minister may, at any time, impose terms and conditions on a medical device licence, or amend those terms and conditions, after considering the following factors:
- (a) whether there are uncertainties relating to the benefits or risks associated with the device;
- (b) whether the requirements under the Act are sufficient to
- (i) maintain the safety and effectiveness of the device,
- (ii) optimize the benefits and manage the risks associated with the device, and
- (iii) identify changes and manage uncertainties relating to the benefits and risks associated with the device;
- (c) whether the proposed terms and conditions may contribute to meeting the objectives set out in subparagraphs (b)(i) to (iii);
- (d) whether compliance with the proposed terms and conditions is technically feasible; and
- (e) whether there are less burdensome ways to meet the objectives of the proposed terms and conditions.
35 Section 37 of the Regulations is replaced by the following:
37 If the terms and conditions of a medical device licence for an in vitro diagnostic device require that tests be performed to ensure that the device continues to meet the applicable requirements of sections 10 to 20, no person shall sell a device from a lot of the in vitro diagnostic device unless
- (a) the results and protocols of the tests performed on devices in the lot have been provided to the Minister; and
- (b) the Minister determines, on the basis of the information provided under paragraph (a), that the devices in the lot continue to meet the applicable requirements of sections 10 to 20.
Regulations Amending the Food and Drug Regulations (Interim Order Respecting the Importation, Sale and Advertising of Drugs for Use in Relation to COVID-19)
36 Paragraph 20(e) of the Regulations Amending the Food and Drug Regulations (Interim Order Respecting the Importation, Sale and Advertising of Drugs for Use in Relation to COVID-19) footnote 36 is replaced by the following:
- (e) sections C.04.004, C.04.005, C.04.007, C.04.009 and C.04.010.
Transitional Provisions
37 Unless the context otherwise requires, the words and expressions used in sections 38 to 43 have the same meaning as in the Food and Drug Regulations.
38 (1) Terms and conditions imposed on a drug identification number under subsection C.01.014.21(1.1) of the Food and Drug Regulations, as that subsection reads before the day on which subsection 6(1) of these Regulations comes into force, that are in effect immediately before that day remain in effect and may be amended by the Minister under subsection C.01.014.21(1.1) of those Regulations as it reads as of that day.
(2) Terms and conditions imposed on a drug identification number under subsection C.01.014.21(1) or (1.1) of the Food and Drug Regulations, as those subsections read before the day on which subsection 6(4) of these Regulations comes into force, that are in effect immediately before that day remain in effect and may be amended by the Minister under section C.01.014.21 of those Regulations as it reads as of that day.
39 (1) The references to “existing plan” in section C.01.702 and subsection C.01.703(1) of the Food and Drug Regulations refer, in the case of a drug for which a risk management plan has been provided to the Minister before the day on which section 7 of these Regulations comes into force, to the most recent version of the plan.
(2) Subsection (1) ceases to apply in respect of a drug when an updated risk management plan is provided to the Minister in respect of the drug in accordance with section C.01.702 or C.01.703 of the Food and Drug Regulations.
40 If, before the day on which subsections 8(2) and 9(2) of these Regulations come into force, an application for or to amend an establishment licence is submitted to the Minister that contains, in accordance with subsection C.01A.005(2) or C.01A.006(1.1) of the Food and Drug Regulations, a statement that refers to COVID-19, the Minister may, on or after that day and despite the amendments made by subsections 8(2) and 9(2), issue or amend the licence on the basis of the application.
41 (1) Subsection C.08.002(2.01) of the Food and Drug Regulations does not apply in respect of a new drug submission that is filed before the day on which that subsection comes into force.
(2) Subsection C.08.003(3.01) of the Food and Drug Regulations does not apply in respect of a supplement to a new drug submission if the supplement is filed before the day on which that subsection comes into force.
42 (1) If, on the day on which subsection 16(1) of these Regulations comes into force, the Minister has yet to make a final decision under section C.08.004 of the Food and Drug Regulations in respect of a new drug submission that was filed before that day in accordance with subsections C.08.002(2) to (2.5) and section C.08.005.1 of those Regulations for a designated COVID-19 drug, as defined in section C.08.001.1 of those Regulations as it reads before that day, subsections C.08.002(2) to (2.5), as they read immediately before that day, continue to apply in respect of the submission until the final decision is made.
(2) If, on the day on which subsection 16(3) of these Regulations comes into force, the Minister has yet to make a final decision under section C.08.004 of the Food and Drug Regulations in respect of a new drug submission that was filed before that day in accordance with subsections C.08.002(2) to (2.5) and section C.08.005.1 of those Regulations for a public health emergency drug, as defined in section C.08.001.1 of those Regulations as it reads before that day, that relates to COVID-19, the drug remains a public health emergency drug for the purposes of Division 8 of Part C of those Regulations until
- (a) the final decision is made; or
- (b) in the case where COVID-19 is described in the List of Conditions that Threaten Public Health, as defined in section C.08.001.1 of those Regulations, when the final decision is made, COVID-19 is removed from that List.
43 (1) If, on or before the day on which subsection 18(7) of these Regulations comes into force, the Minister indicates to a manufacturer of a new drug for veterinary use only who has filed a new drug submission for that drug that does not yet contain all of the information and material that is required to be contained in the submission an intent to conduct a review of the submission with a foreign regulatory authority, the manufacturer must, within 60 days after that day, provide the Minister with a plan that specifies how and when they will provide the Minister with the missing information or material, unless they have provided the Minister with such a plan on or before that day.
(2) If, on or before the day on which subsection 21(3) of these Regulations comes into force, the Minister indicates to a manufacturer of a new drug for veterinary use only who has filed a supplement to a new drug submission for that drug that does not yet contain all of the information and material that is required to be contained in the supplement an intent to conduct a review of the supplement with a foreign regulatory authority, the manufacturer must, within 60 days after that day, provide the Minister with a plan that specifies how and when they will provide the Minister with the missing information or material, unless they have provided the Minister with such a plan on or before that day.
44 Any terms and conditions that, immediately before the day on which section 34 of these Regulations comes into force, are set out in a medical device licence referred to in subsection 36(1) of the Medical Devices Regulations are deemed to be imposed by the Minister under subsection 36(2) of those Regulations, as that subsection reads on that day.
Coming into Force
45 (1) Subject to subsections (2) and (3), these Regulations come into force on the day on which they are registered.
(2) The following provisions come into force on the one-year anniversary of the day on which these Regulations are registered:
- (a) sections 4 and 5;
- (b) subsection 6(4);
- (c) section 7;
- (d) subsections 18(2), (5) and (7);
- (e) sections 19 and 20;
- (f) subsections 21(1) and (3);
- (g) section 23; and
- (h) sections 34 and 35.
(3) The following provisions come into force on a day to be fixed by amendment to this subsection:
- (a) subsections 8(2), 9(2) and 10(2);
- (b) subsection 16(3); and
- (c) subsections 18(4) and (6).
Terms of use and Privacy notice
Terms of use
It is your responsibility to ensure that the comments you provide do not:
- contain personal information
- contain protected or classified information of the Government of Canada
- express or incite discrimination on the basis of race, sex, religion, sexual orientation or against any other group protected under the Canadian Human Rights Act or the Canadian Charter of Rights and Freedoms
- contain hateful, defamatory, or obscene language
- contain threatening, violent, intimidating or harassing language
- contain language contrary to any federal, provincial or territorial laws of Canada
- constitute impersonation, advertising or spam
- encourage or incite any criminal activity
- contain a language other than English or French
- otherwise violate this notice
The federal institution managing the proposed regulatory change retains the right to review and remove personal information, hate speech, or other information deemed inappropriate for public posting as listed above.
Confidential Business Information should only be posted in the specific Confidential Business Information text box. In general, Confidential Business Information includes information that (i) is not publicly available, (ii) is treated in a confidential manner by the person to whose business the information relates, and (iii) has actual or potential economic value to the person or their competitors because it is not publicly available and whose disclosure would result in financial loss to the person or a material gain to their competitors. Comments that you provide in the Confidential Business Information section that satisfy this description will not be made publicly available. The federal institution managing the proposed regulatory change retains the right to post the comment publicly if it is not deemed to be Confidential Business Information.
Your comments will be posted on the Canada Gazette website for public review. However, you have the right to submit your comments anonymously. If you choose to remain anonymous, your comments will be made public and attributed to an anonymous individual. No other information about you will be made publicly available.
Comments will remain posted on the Canada Gazette website for at least 10 years.
Please note that public email is not secure, if the attachment you wish to send contains sensitive information, please contact the departmental email to discuss ways in which you can transmit sensitive information.
Privacy notice
The information you provide is collected under the authority of the Financial Administration Act, the Department of Public Works and Government Services Act, the Canada–United States–Mexico Agreement Implementation Act,and applicable regulators’ enabling statutes for the purpose of collecting comments related to the proposed regulatory changes. Your comments and documents are collected for the purpose of increasing transparency in the regulatory process and making Government more accessible to Canadians.
Personal information submitted is collected, used, disclosed, retained, and protected from unauthorized persons and/or agencies pursuant to the provisions of the Privacy Act and the Privacy Regulations. Individual names that are submitted will not be posted online but will be kept for contact if needed. The names of organizations that submit comments will be posted online.
Submitted information, including personal information, will be accessible to Public Services and Procurement Canada, who is responsible for the Canada Gazette webpage, and the federal institution managing the proposed regulatory change.
You have the right of access to and correction of your personal information. To seek access or correction of your personal information, contact the Access to Information and Privacy (ATIP) Office of the federal institution managing the proposed regulatory change.
You have the right to file a complaint to the Privacy Commission of Canada regarding any federal institution’s handling of your personal information.
The personal information provided is included in Personal Information Bank PSU 938 Outreach Activities. Individuals requesting access to their personal information under the Privacy Act should submit their request to the appropriate regulator with sufficient information for that federal institution to retrieve their personal information. For individuals who choose to submit comments anonymously, requests for their information may not be reasonably retrievable by the government institution.